
Ensuring source material consistency & continuity for commercialization of advanced therapies
Cell & Gene Therapy Insights 2020; 6(2), 295-305
10.18609/cgti.2020.040
A critical aspect to ensuring patient access to cell and gene therapies (CGT) and continued growth of the industry is having a proper awareness for managing the source material quality and supply chain continuity. The combination of rapid growth, individual product and process complexity, and limited industry-specific guidance or awareness presents ongoing challenges for transitioning from development to clinical and commercial manufacturing scale. For allogeneic therapies, having access to consistent and reliable donors and high quality, GMP-compliant starting material, coupled with the ability to consistently deliver this clinical source material to the required point of use, will be key to long-term success.
Introduction
The cell and gene therapy industry continues to grow and progress at an exciting rate [1]. More and more products are advancing from research and preclinical development into the clinic. However, it is also very much a pivotal period, as we look to venture to the next phase of industry growth and maturity: as clinical successes translate to commercialization, bioproduction resource demand has the potential to reach unprecedented levels [2].
There are notable aspects to this expected trend. Firstly, it is important to realize that the ever-increasing number of clinical trials that we are so accustomed to seeing remains just a small fraction of the anticipated demand. Secondly, despite the fact that autologous products will continue to build from their early success, it will be allogeneic therapies and products that will really drive future growth and resource demands in the cell therapy arena. (Figure 1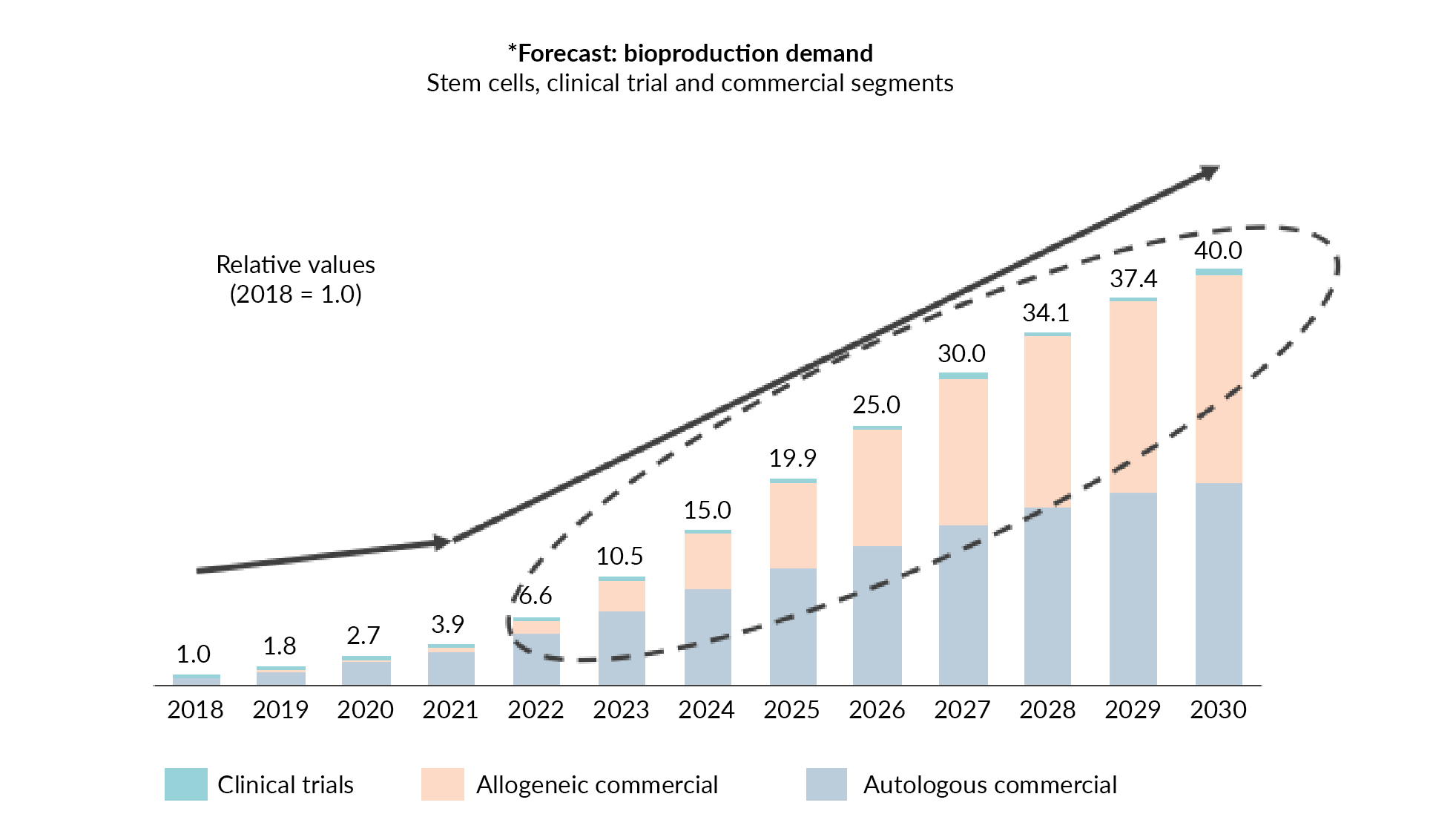
Over the next 5 years, we could be experiencing a 15–20-fold bioproduction growth in the cell therapy segment. This type of growth requires continued industry collaboration and innovative strategies. A key component in accommodating the forecasted resource demands is developing a reliable and consistent supply of critical raw materials including therapeutic starting materials.
Contributing factors to consider for donor-derived starting material
Donor starting material is traditionally a necessary component for development of both autologous and allogeneic cell therapies. It can be easy to take something complex like cellular starting material and simplify it, especially when the industry progresses through development at such a rapid pace. However, taken from a different perspective, the actual starting material used in downstream processing is a complex combination of different components, including donor management, physical collection, processing steps, and shipping. Understanding and managing each of these contributing factors will help to achieve a more consistent and sustainable source – especially in the context of anticipated industry scale-up demands.
Any cell therapy process typically becomes a balancing act between meeting the immediate, early bioprocess development needs versus the clinical and commercial requirements: short-term versus long-term considerations; highly variable versus highly controlled processes; small-scale versus large-scale; research use versus full GMP compliance (or feasibility versus safety and consistency). Given this balancing act, it’s easy to see why development practices can hide potential quality, sourcing, and logistical challenges that will be faced eventually with the clinical translation of cell and gene therapy products. The same is true for the starting material. Considering donor management as an example, it is critical to factor in the impact of eligibility and recruitment – not only for safety but also availability – on collection and processing needs.
With cellular starting materials, we often think immediately about the variability. Given the nature of these therapies, variability may be a desirable trait at times – when it can be controlled, of course – but quality and compliance become major factors as the therapeutic product progresses into and through the clinic. Finally, shipping (as discussed in greater detail below) is always critical. The inherent stability challenges with cellular products require careful consideration early in development, as they will impact availability. But one must also be aware of a product’s packaging and transit needs and ultimately, it’s traceability – the assurance that the material will arrive where it needs to be, when it needs to be there, and in the specified condition.
Donor management & eligibility
Donor management is a central aspect to any normal healthy donor-derived starting material. From a regulatory perspective, donor eligibility requirements are very well defined with screening and testing designed to minimize the risk of infectious disease transmission. Box 1 lists the standard tests required, which are generally aligned across the major regulatory bodies (e.g. the US FDA and EMA). Unfortunately, full global regulatory alignment does not currently exist [3,4]. Therefore, care must be taken, especially for material being used for allogeneic purposes and the development of cell banks [5]. Awareness and traceability are always important, particularly when considering how to manage new disease concerns like COVID-19 for example.
Less well defined, but equally critical, are the eligibility or suitability criteria for the specific clinical protocol defined by the sponsor. During development, one may set restrictive limits around age or body mass, for example. Some stringency may be required, but these restrictions can significantly reduce the preferred donor pool resulting in challenges to donor or product access. Therefore, developing and implementing a robust donor strategy with the starting material supplier is needed.
| Box 1: Standard infectious disease testing |
|---|
|
Managing starting material variability
While donor management is a key aspect of both safety and supply continuity, perhaps the most commonly referenced challenge is that of starting material variability. Figure 2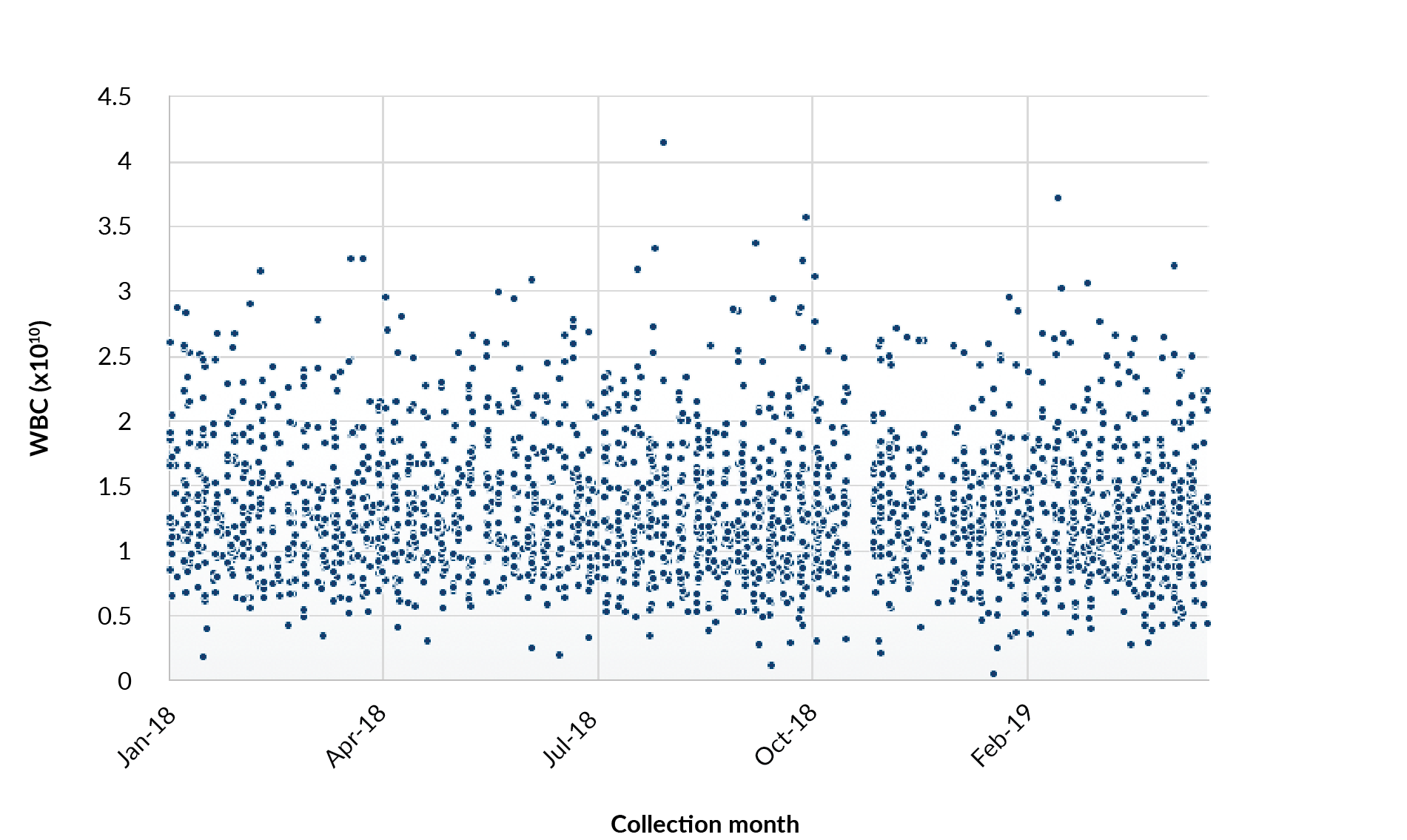
For allogeneic therapies where the donor starting material is used for the final product, variability can also be problematic for manufacturing consistency. Regardless of the product type, developing a robust method or strategy to address variability is critical.
Having access to reliable and recallable donors is a highly effective method for addressing starting material variability and developing a robust supply chain. Examining cellular subsets of reliable and recallable donors, reveals an additional key benefit. A certain process may be reliant on having specific biological characteristics, such as a high percentage of CD3 or CD4 positive cells. By harnessing data from repeat donors, developers may obtain the starting material that best fits their manufacturing requirements. This leads to improvements in the management of starting material variability and thus, to an improved process and final product consistency (Figure 3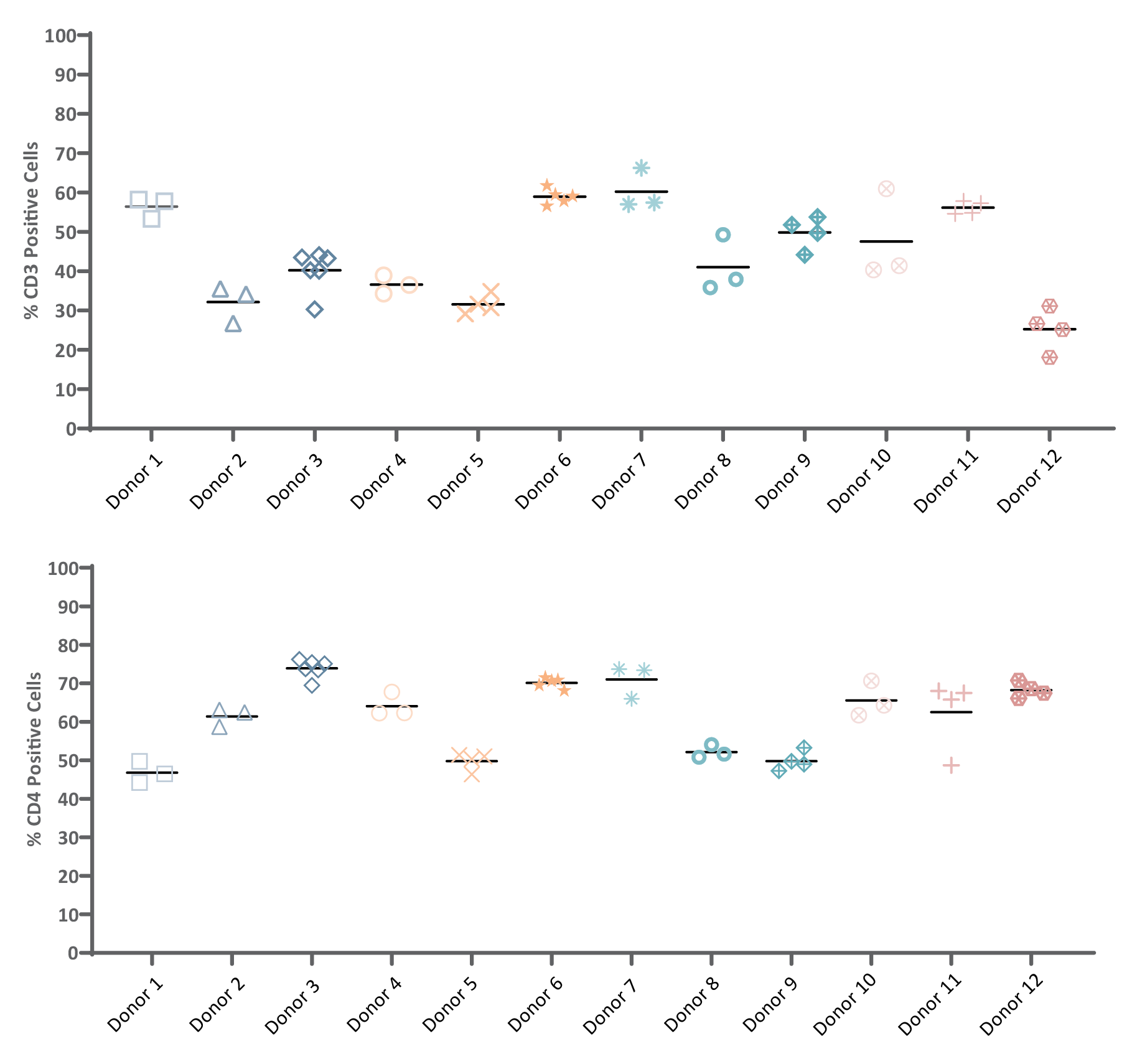
Quality/stability
Obtaining the starting material and managing its inherent variability are challenging in their own right, but given the nature of biologicals, a keen awareness of the overall product stability is also necessary. Often in early process development, starting materials are collected and shipped fresh for downstream processing. Fresh, cell-based starting materials have limited shelf-lives. The potential loss in starting material quality and stability over time further exacerbates the variability and can greatly impact process and product consistency due to overall cell loss, functional limitations, or both. Furthermore, the stability limitations require the collection and shipping to occur without delay. This presents a challenge for all involved as delays will inevitably occur, and the level of risk only increases with scale [6].
Optimizing post-collection processing and shipping requirements early in development is necessary to maintain starting material quality, and a clearly defined strategy in this regard will be essential for long-term clinical and commercial efficacy of the final therapeutic product. Cryopreservation of the starting material, whether it is the entire leukapheresis or an isolated sub population of cells, can reduce or eliminate the stability limitations. However, the success of such a strategy is in part dependent on working with a partner that has the resources and capabilities to perform these steps onsite and immediately post-collection, in order to maintain the highest potential quality. The appropriate stability needs to be maintained at scale, so defining an end-to-end starting material strategy will go a long way towards simplifying donor management and collection demands, thus ensuring appropriate staffing and scheduling at both the collection site and the downstream manufacturing site. It is also ideal for all parties to work with a logistics partner that can manage the considerable shipping demands involved in enhancing product consistency and integrity, which range from the pack-out and handling of materials for process development, to meeting the critical compliance requirements of the starting materials for clinical and commercial use.
| Box 2: Focus on packaging |
|---|
| Not only does packaging physically protect the material from a variety of external factors, it also maintains the correct environment, can facilitate use, and most likely also includes some aspect of recording and/or location device. Various risk factors can affect packaging performance and consistency, from the basic design of the packaging itself to its preparation, handling, adherence to a specified pack-out, and even how the source material itself is provided – for example, warm fresh material that is to be maintained at a refrigerated temperature. As products move to GMP supply chains, the associated packaging often needs to be demonstrated and documented as appropriate, as part of the developer’s GMP obligation. In most cases, a commercially available temperature-controlled package is utilized as the best option. This provides a high performance, secure and cost-effective method of protecting the product during transit, as well as being appropriately scalable. This packaging may contain bespoke elements – for example, product-specific inserts to provide additional protection or aid use – and have completed general qualification and validation by the vendor and service provider. With the move to clinical and commercial products and associated GMP requirements, the drug developer is obligated to identify what qualification and validation work is needed to prove control of the critical aspects of their operation. Individual products will each likely have a specified stability and associated transport temperature range, as well as a defined primary or secondary packaging type and routing profile. Vendor and service provider validation reports will often be based on generic product loads and temperature ranges. In extreme cases, these may differ greatly from the specific developer’s needs based upon expected product load/volume/temperature range. Ultimately, it is the drug developer’s responsibility to determine what is acceptable under their GMP obligation and this may require additional specific testing to be completed. Before completing any specified testing, a detailed protocol should be agreed and approved to make sure that the resulting report is accepted. When designing the protocol, consideration should be given to the expected usage and worst-case-scenario options should be identified and tested. For instance:
When completing testing, it is vital that documented processes are followed so that a true reflection of expected use is measured. When creating the summary report, any deviations from the protocol must be clearly recorded and documented along with detailed results and measurements taken during the test. |
Keys to translational success
As previously mentioned, the starting material needs for development purposes are often not completely aligned with the clinical requirements. To define a strategy for translational success, it is important to consider all aspects of the starting material – including donor management, collection and processing, and shipping – from an early stage of development, especially for allogeneic cell products.
Clinical translation begins with donor management and careful planning around donor eligibility. Knowledge of the intended clinical regions will help ensure that appropriate screening and testing for regulatory compliance is performed prior to collection, and also for any long-term cell banking plans. A further important consideration for consistent starting material access relates to including potentially excessive restrictions, which can result in significant limitations. Access to well characterized, reliable and recallable donors can alleviate some of the risks associated with donor management.
The collection and processing requirements of the starting material for clinical use are critical to all cell and gene therapy products. For any immediate post-collection processing of the starting material, the facilities and processes need to be qualified and validated for GMP-compliant manufacturing. For allogeneic therapies, working with a starting material provider that can perform the GMP-compliant collection and processing onsite eliminates risk and provides greater consistency.
| Box 3: Case Study: Beam Therapeutics |
|---|
| Beam Therapeutics is developing precision genetic medicines through base editing. Unlike other modalities of genetic engineering, Beam’s novel approach to gene editing allows the replacement of single base pairs without initiating double-stranded breaks in DNA; if other gene editing platforms can be considered to act as scissors, Beam’s platform is more akin to a pencil and eraser. The double-stranded break mechanism of editing can result in nonhomologous end joining in the DNA strand, which leads to cell disrepair and death. However, Beam’s technology allows for a narrow focus on a single base pair, and the ability to change that base pair without disturbing the integrity of the DNA. Beam is applying this technology in a wide array of indications. However, this case study will focus on the company’s two leading cell therapy platforms: in the autologous setting for patients with sickle cell disease and beta thalassemia, and in the allogeneic setting for patients with leukemia. The cell therapy supply chain is a new and unique supply chain that presents its own challenges. For autologous therapies, cells are collected, shipped to manufacturing sites, processed into drug products, and shipped back to the patients. Depending on a given therapy’s manufacturing site, cells may need to be shipped globally both from and then back to the patient, leading to the requirement for timely, robust, and traceable logistics. Overall, the allogeneic therapy supply chain is similar to that of autologous products. Instead of the patient’s cells being collected, a healthy volunteer donates cells to be shipped to the manufacturing site. Those cells are processed, and the drug product may then be shipped to multiple patients in need. Although one element of the supply chain is eased through the collection of cells from a single donor, the dependence on a living start material still exists: the quality of this material could be compromised easily by low quality collection, delayed shipment, or reduced viability of the cell product. Beam’s autologous hematological disease programs rely on fresh, mobilized apheresis. While the composition of the leukopak will differ between healthy donors in development and patients during clinical trials, we have identified the dosing regimen required to achieve the starting population needed for development. Beam is leveraging HemaCare’s expertise to improve the definition of our collection requirements for our clinical trials. Meanwhile, to maximize our ability to address the challenges surrounding the shipment of fresh apheresis, we are working with Biocair as our shipping logistics and chain of custody partner for this program. Biocair has reliably delivered these living products across the US without diminishing their quality. We’ve benefited from the real-time tracking of shipments that Biocair provides. For example: recently, a leukopak was shipped to our contract manufacturing organization. However, after the certificate of analysis was released, we realized the cell count was too low for our intended purposes there. We were able to update the shipping address in real time to ensure the product was delivered to our process development lab instead. Despite this re-routing, Biocair still managed to deliver the leukopak before 9.00 a.m. Beam has leveraged HemaCare’s GMP pipeline for apheresis, identifying donors who met our collection requirements and performed well in our research process, and banking them for manufacturing. The scope of the GMP platform for apheresis collection has been critical to our campaign, allowing us to find strong donors whom we can fully characterize in process development, and then have them recollected in the GMP setting at a later date for manufacturing purposes. In order to have a large pool of donors to select from, we started with minimum donor criteria. As we received input from clinicians, we then narrowed the donor criteria, which means that consistent screening of our selected donors is required. These requirements of our selected donor pool, along with the ever-present possibility that a donor could be sick or ineligible, or drop out of the donor pool completely, are concerning in the long-term. To overcome this issue, we aim to create a donor pool that is twice the size of that which we anticipate needing. To further secure our supply of starting material, we’ve built our manufacturing process around frozen apheresis, thus securing the front end of the supply chain and ensuring we can access apheresis at any time from our selected donor bank. As the cell therapy field continues to grow, we expect regulatory guidance to evolve. We will need to continue to adapt to the input of regulators as this occurs and because of this, we will need to keep strong records of our previous donors. This will in turn allow us to keep our starting material and our lots of already produced, ‘on the shelf’ drug product compliant with regulation. Collecting leukopak with batch records and well-documented processes will help us as developers to keep the appropriate records of our donor pool, to hopefully keep ahead of growing guidance, and ensure evolving safety measures for our patients. In cell therapy, the largest source of variation is the donor. Variability in the apheresis collection yields variation in the manufacturing process, which yields variation in the drug product. With allogeneic therapies, we can overcome some of this issue by pre-screening donors against our defined critical quality attributes. Unlike autologous therapies, where manufacturing is at the mercy of the incoming starting material, there’s an opportunity to treat apheresis like any other starting material and find the best suited donor for your process. Beam has utilized HemaCare’s network of recallable donors to bring in a variety of donors, test them against key process parameters, and assess their overall quality within our system. Once identified, we can request those donors be put on hold for drug product manufacturing. We have tested both fresh, and frozen apheresis, and have found that for some key readouts, there is no impact from beginning with frozen material: there is no difference in the starting viability of our incoming cell product, whether it has been shipped fresh or thawed from frozen apheresis. Furthermore, post-processing, we maintain that same high viability regardless of whether the starting material was fresh or frozen. These results, and other critical quality attributes we have determined, led to the implementation frozen starting material for our development and manufacturing processes. The inclusion of frozen apheresis has also allowed us to bank our selected donors and save them for future manufacturing campaigns, further securing our supply chain. From a development perspective, we believe our best path forward towards enabling the successful manufacture of Beam’s cellular therapies relies on building a process around robust starting material. It is in the developer’s best interest to define collection procedures and to minimize incoming starting material variation, regardless of whether the therapy is autologous or allogeneic. Particularly in allogeneic therapies, we can build processes around high quality, frozen apheresis to secure the supply chain and allow for preservation of starting material that is specific to our process. Lastly, Beam feels it is important to take learnings from the industry by using validated shipping methods that preserve temperature- sensitive starting materials and provide chain of custody, so that the manufacturing process has the greatest chance of success. Through these learnings we can guarantee that the best possible drugs are getting to patients in need. |
Delivering logistical continuity of source materials for commercialization of advanced therapies
Shipping represents the final piece of the puzzle and although various options exist for managing the shipment of GMP-compliant starting materials, ultimately having a trusted logistics partner is key to success.
It is vitally important to properly consider the impact that logistics may have on the success of each new advanced therapy, particularly as products move from early development firstly to clinical and then to commercial scale. As volumes and scale increase, any weaker points in the supply chain are liable to become more exposed and therefore, an awareness of the impact on the required logistical solutions for the supply of source material is key to allowing the correct planning to be undertaken. This awareness may assist in decisions around the supply of frozen or fresh material, planning the timing and location of donor visits, or even manufacturing scheduling or location.
Maintaining consistency as scale and associated volumes increase is essential. Throughout this transition and on an ongoing basis, risk must be identified and effectively managed. The key areas of risk are likely to evolve throughout this transition, in line with the increase in scale and volume, and it should not be assumed that the same solutions should be rigidly adopted throughout. Some high-level areas that require effective control include traceability, packaging, routing, as well as regulatory and quality assurance.
GDP is the legal standard for logistics companies distributing medicinal products to ensure their safety, quality, efficacy, and traceability throughout the supply chain. GDP controls many areas from Quality Management, Premises & Equipment, Documentation and Record Keeping, through to Change Control, Cold Chain, and Training. Specialist logistics providers should already be operating to GDP as standard, providing reassurance of preparedness for the move away from the development stages. Where non-specialist / non-GDP providers have been utilized, significant additional planning and change should be anticipated.
Although a specialist logistics provider should already operate to GDP, some further controls also now need to be considered: GDP sets out the framework and requirements but should be seen as a minimum when moving GMP starting material, as additional control and knowledge are often needed. The differing requirements of individual products can require a tailored approach to effectively manage risk.
As well as effective risk management, there are certain aspects that are key in delivering continuity at any stage. Firstly, a clear understanding of ongoing needs is vital to provide the optimal solution from the outset. This may involve specific product sensitivities, controls or impacts on the donor, medical professionals, or manufacturer. While the core specialist logistical service may provide a 99% solution, the additional 1% tailored to the specific product can make all the difference, from controlling the packaging, through regulatory knowledge and production of associated paperwork, to the retrieval of temperature results. By taking total ownership of the whole process, the logistics provider is not only able to allow you to focus on your core tasks (and not logistics) but also better able to manage the transition from development.
As volumes increase, finding an intuitive solution that is easy to operate at each touchpoint is essential. From collection site to use at the clinic, there is a balance to be found between complexity and risk. Training and control through a quality system with effective CAPA management and suitable quality culture is another key aspect, ensuring that the whole network is working to the same processes. Technology must be utilized correctly as volumes grow, removing risk of human error where possible, but simpler solutions should not always be immediately dismissed.
Ultimately, a logistics provider must be trusted to safely collect, transport, and deliver to the correct place on time. These fundamentals must not be overlooked when managing and planning for increasing scale.
In summary, the importance of considering the impact that logistics will have and how this could influence decisions as scale increases must not be overlooked. Of further importance is ensuring the effective control of risk, bearing in mind that the major risk areas may change and evolve as a product progresses towards commercialization. Maintaining total ownership of processes facilitates management and control by aligning each step from packaging and collection though to final delivery is strongly recommended, as is involving a logistics specialist in order to help navigate and understand the impact of key decisions.
Conclusion
To help smooth the transition to commercialization for cell therapy developers, HemaCare and Biocair are pooling knowledge and developing universal solutions for the supply of GMP materials to the point of required use. The aim of this is to:
- Facilitate the move to GMP supply chains
- Provide clear pathways for managing increasing scale
- Limit additional workload of developers through product-specific solution qualification and validation
- Ensure the consistent and timely supply of starting materials throughout the lifecycle of any given product

Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The authors declare that they have no conflicts of interest.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2020 Hemacare. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited. This article has been created from a previously aired webinar that can be found here.
Submitted for peer review: Mar 12 2020 Publication date: Apr 8 2020.
References
1. Alliance for Regenerative Medicine. 2019 Annual Report and Sector Year in Review: Website
2. Taylor B, Clarke D. The Evolving Role of Starting Materials in Cell and Gene Therapies. BioPharm Int. 2019; 32(12): 30–5. Website
3. Read E. Qualification of cellular starting materials for cell-based therapies. Cell Gene Ther. Insights 2019; 5(2): 177–96. CrossRef
4. Bravery B, Robinson S, Burger S. Making the grade: untangling the myths of raw materials used for the manufacture of cell- and gene-based medicinal products. Cell Gene Ther. Insights 2018; 4(3): 207–25. CrossRef
5. Magers K. Addressing Challenges in Meeting Chemistry, Manufacturing and Control Regulatory Requirements for Gene Therapy Products. Cell Gene Ther. Insights 2019; 5: 1–16. Website
6. Clarke D, Smith D. Managing Starting Material Stability to Maximize Manufacturing Flexibility and Downstream Efficiency. Cell Gene Ther. Insights 2019; 5(2): 303–13. CrossRef
Affiliations
Dominic Clarke
Global Head of Cell Therapy, Hemacare
Christopher Good
Director, Cell & Gene Therapy Logistics, Biocair
Amy Shaw
Lead, Cell Therapies Engineering and Automation Development at Takeda (Formerly: Senior Scientist, Cell Therapy Development, Beam Therapeutics)
![]()
![]()