Challenges of developing anti-drug antibody & concentration (PK or PD) assays for liposomal transgene enzymes on an automated immunoassay platform
Cell & Gene Therapy Insights 2023; 9(3), 241–251
DOI: 10.18609/cgti.2023.036
Gene therapy can be used to permanently correct genetic disorders by delivering a transgene product into the nucleus of affected or alternative cells. One of the biggest challenges faced by the gene therapy bioanalytical sector is the lack of a true reference material for transgene products. As a result, in most assays, an alternative commercially available therapeutic (for example, an enzyme replacement therapy product) can be used as a surrogate for the transgene enzyme. This article discusses approaches taken in order to monitor the concentration of the expressed transgene product and any associated immunogenicity.
Transgenes are expected to have very low levels of immunogenicity; therefore, immunogenicity assays against transgenes themselves are not widely developed. Nonetheless, it is important to monitor any potential case of immunogenicity. Current regulatory guidelines are not applicable to transgene products, which adds to the challenge. The following case study describes the considerations and challenges encountered when developing and validating an assay for transgene lysosomal enzyme products. Since enzymes are sensitive to changes in pH and salt, specific buffers were required to maintain their optimal configuration to ensure all types of anti-drug antibodies (ADA), including neutralizing antibodies (Nabs), are detected.
The same level of challenges was also encountered during the development of an assay to determine the transgene product concentration, mainly due to the lack of a true reference standard and the presence of the equivalent endogenous molecule in healthy and disease matrices. Therefore, measurement of the transgene concentration could not be described as a true pharmacokinetics (PK) assay and does not conform to current the PK guideline/guidance, requiring an approach more aligned with a pharmacodynamics (PD) assay with a well-defined context of use.
The Gyrolab® platform for immunoassays
Gyros Protein Technologies has supplied the market with proprietary high-performance nanoliter-scale immunoassay platforms, used by scientists in leading pharmaceutical, biotech, contract research organization, and contract manufacturing organization companies since the beginning of 2000 [1].
The core of Gyrolab technologies includes the range of compact discs (CD), which are the site of the immunoassay reaction, or the equivalent of a microtiter plate, involving precise volume definitions. Samples and reagents are transferred to the 15 nL affinity flowthrough column, which allows various assay formats. Detection involves laser-induced fluorescence to give an indication of assay binding. This provides good assay sensitivity in addition to a broad dynamic range.
The Gyrolab immunoassay platform is fully automated and has an integrated workflow to enable increased productivity and reproducibility. Volumes are at a nanoliter scale to save both reagent and sample. The platform enables short turnaround times, with a full assay taking approximately 1 h to perform. The platform’s broad dynamic range reduces the number of dilution steps in the assay. Reduced matrix effects facilitate assay transfer in all stages of drug development.
The open platform results in a flexible approach supporting many different assay designs, including sandwich, PK, bridging, and indirect assay formats in one-, two-, three-, four- or five-step processes. Gyrolab methods supporting different assay designs are available for download.
An overview of AAV gene therapy bioanalysis
Adeno-associated viruses (AAV) are replication-deficient, non-enveloped viruses. They hold promise for use in gene therapy due to their low immunogenicity, and their demonstrated long-term gene expression. AAV can transduce a wide variety of tissues, with over 11 well-known human viral serotypes. AAV are not known to cause any human diseases. Their mode of action is described in Figure 1 Adeno associated virus mode of action.AAV; Adeno associated virus: ADA; Anti-drug antibodies; Nabs; Neutralizing antibodies; PD; Pharmacodynamic: PK; Pharmacokinetic..
Adeno associated virus mode of action.AAV; Adeno associated virus: ADA; Anti-drug antibodies; Nabs; Neutralizing antibodies; PD; Pharmacodynamic: PK; Pharmacokinetic..
In terms of bioanalysis, one key factor is to understand if there is anything that prevents AAV from entering cells i.e., pre-existing antibodies against AAV. Through assay development, it has been demonstrated that in some cases, over 70% of pre-existing antibodies are present for some types of AAV. It is important to pre-screen to avoid the presence of those candidates in a trial, or for the removal of those pre-existing antibodies.
Once the AAV is inside host cells, the genome material from the AAV needs to be monitored, usually by qPCR.
Transgene quantification
The quantification of the transgene does not follow current PK guidelines. The lack of a true reference standard is the main challenge, meaning that the only quantification that can be done on a transgene product is relative. To quantify a product, an alternative available therapy that mimics expected transgene must be used as a calibrator.
Due to this, the quantification of transgene products is more aligned with biomarker assessment, or PD. As a result, the context of use is more important for transgene assessment. For example, there may be an interest in looking for the change from baseline to physiologically relevant levels.
An important factor to consider is that expressed transgene product will closely resemble the endogenous protein, and it can be difficult to distinguish them. Liquid chromatography–mass spectrometry (LC–MS) is advisable if the expressed protein has a unique structure different from that of the endogenous counterpart. However, the LC–MS method might lack some of the sensitivity that a ligand-binding assay might provide. In every case, parallelism and stability are crucial. Therefore, the assay is only fit for purpose and only becomes relevant once parallelism has been demonstrated in the sample containing transgene product.
Key considerations for transgene quantification include sensitivity, which can be as low as the nanogram or picogram range. However, transgene expression is expected to be consistent, so the required dynamic range of an assay is small. A stepwise format is the standard go-to for most quantitative methods. However, in cases of transgene products where the assay range is not large, a homogenous assay format can be accepted. In the examples described here, a homogenous assay format was demonstrated to provide the best sensitivity, and the best parallelism compared to a sequential format.
When working with transgene enzymes, it is important to optimize the buffer, as enzymes can be prone to conformational change. Conformational change can lead to epitope-masking, resulting in poor recovery (Figure 2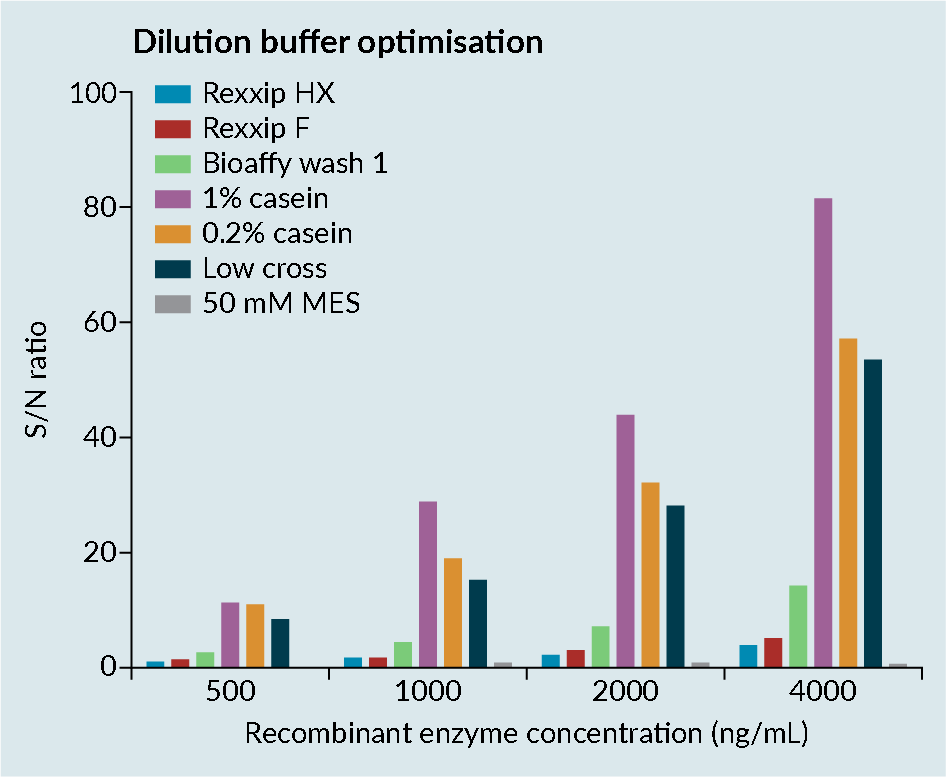 Enzyme optimization consideration.).
Enzyme optimization consideration.).
Figure 2 shows the dilution buffer optimization assessment of a transgene enzyme in various buffers, conducted by the contract research organization Drug Development Solutions.
Rexxip™ buffers, which are known to work well for antibodies, were demonstrated to not be compatible with enzymes, likely due to the buffer salt content. Simple buffers such as casein improved the sensitivity of the assay. However, casein has poor solubility and the precision in the assay was
relatively poor.
When developing an assay, it is important to consider temperature, pH, and salt concentration, which are especially vital when using enzymes, as they can play a key factor in conformational change. The treated matrix was prepared by heating to 56°C to denature the endogenous enzyme. In this case, heat-treated matrix improved parallelism compared to the use of any other surrogate matrix.
Platform comparisons were performed to evaluate the sensitivity and precision of Meso Scale Discovery (MSD) and the Gyrolab platform (Figure 3 Platform comparisons: MSD versus Gyrolab platform sensitivity and precision.CV; Cell viability: MSD; Mesoscale discovery; S/N; Signal-to-noise; QC: Quality control.). It was demonstrated that the Gyrolab offered slightly improved sensitivity and precision compared with MSD. In addition, the Gyrolab gave much better parallelism data, compared to the MSD (Figure 4
Platform comparisons: MSD versus Gyrolab platform sensitivity and precision.CV; Cell viability: MSD; Mesoscale discovery; S/N; Signal-to-noise; QC: Quality control.). It was demonstrated that the Gyrolab offered slightly improved sensitivity and precision compared with MSD. In addition, the Gyrolab gave much better parallelism data, compared to the MSD (Figure 4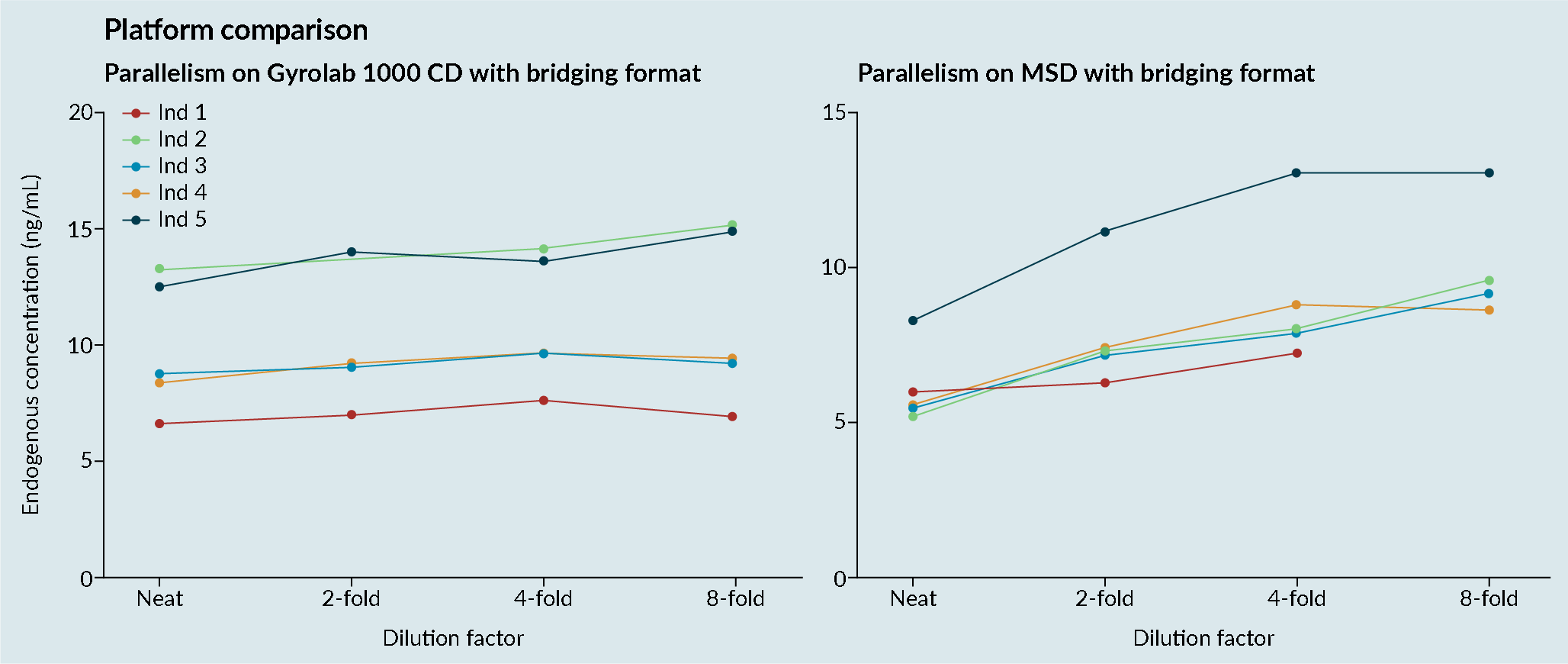 Platform comparison: MSD versus Gyrolab platform parallelism.). This led to the work being taken forward on the Gyrolab. As discussed previously, parallelism of the endogenous enzyme was crucial to demonstrate that a ligand-binding assay method is fit for purpose.
Platform comparison: MSD versus Gyrolab platform parallelism.). This led to the work being taken forward on the Gyrolab. As discussed previously, parallelism of the endogenous enzyme was crucial to demonstrate that a ligand-binding assay method is fit for purpose.
ADA method assessment
As with the PK assay, there is no current guidance on immunogenicity assessments of transgene products. The current FDA guideline clearly stipulates that it does not relate to cell and gene therapy products. While developing these methods, it was necessary to look elsewhere to establish the most appropriate means to assess the immunogenicity of
transgene products.
At Drug Development Solutions, the same approach has been adopted for assessing anti-transgene product antibodies as for a normal protein (Figure 5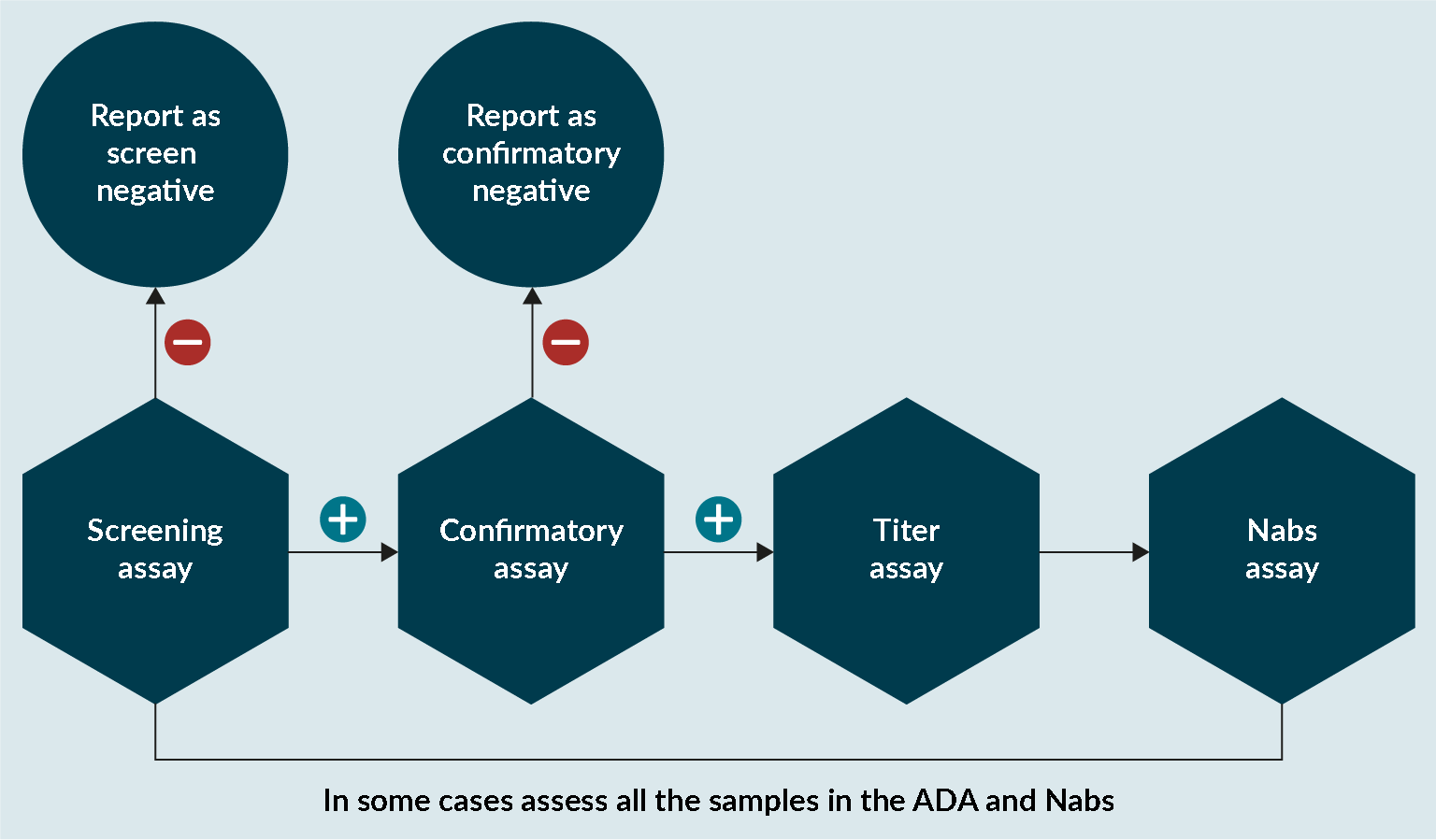 An approach to assess anti-transgene product antibodies.). In some cases, it is important to assess if conformational changes have affected the enzyme and that the catalytic unit is not affected. If there is any evidence that this is the case, then it is important that any sample analyzed in the ADA assay is also analyzed in a NAbs assay.
An approach to assess anti-transgene product antibodies.). In some cases, it is important to assess if conformational changes have affected the enzyme and that the catalytic unit is not affected. If there is any evidence that this is the case, then it is important that any sample analyzed in the ADA assay is also analyzed in a NAbs assay.
Prior to ADA assay development, it was known that the transgene endogenous circulating concentration was going to be present at a very low level – this provided the flexibility to be able to develop a homogenous or stepwise assay format. Both formats would have required the need to conjugate the enzyme. Enzyme conjugation provides additional challenges such as the need to be kept in the right medium, to avoid epitope masking.
Method development was initially carried out using the Gyrolab xPlore, and transferred to the Gyrolab xP or xPand as they allow the use of more CDs. When the conditions were selected, the capture was dissolved in Rexxip F, the detection was diluted in Rexxip F, and the solution was diluted five-fold in Rexxip ADA. When the method was transferred, an assessment of multiple CDs demonstrated loss of the assay signal.
To investigate what caused these issues, Drug Development Solutions approached Gyrolab to assist in determining which buffer was appropriate. The first point of call was looking at a buffer with an isoelectric point closer to the enzyme of interest. With the Rexxip Hx, a similar signal deterioration over multiple CDs was found, with high coefficients of variation (CVs) more than 20% also observed. It was apparent throughout these experiments that the loss of signal was also associated with higher precision failure.
Given that the buffers were the root cause of this problem, the simplest buffer from Gyrolab, the wash station solution 01, was then assessed. This demonstrated a stable signal; however, the baseline was high, leading to poor sensitivity. The increase in signal also improved the CVs. These data clearly demonstrated that the Rexxip buffers were causing issues with the enzyme, which helped in terms of optimizing other buffers.
In this case, casein was found to improve assay sensitivity and reduce the baseline to the desired levels. However, precision was an issue for casein. The Gyrolab team advised increased centrifugation and an additional mixing step to improve the precision issue, which was successful.
Conclusions & recommendations
It is crucial to consider that enzymes are especially sensitive to temperature, salt, and pH change. Care is required to avoid missing a positive result as a result of enzyme conformational change. Where possible, assess conjugated enzymes using a potency/NAbs assay to confirm activity. Before transferring from the Gyrolab xPlore to the Gyrolab xP, extended sample stability and extended reagent stability should be assessed. All CDs should be spun simultaneously to reduce challenges with reagent and sample stability and shorten the run time.
Reference
Gyros Protein Technologies. Installations worldwide. Crossref
Ask the Authors


Issa Jyamubandi, Principal Scientist, Alliance Pharma and John Chappell, Director of Scientific Support, EMEA and Asia Pacific,Gyros Protein Technologies (pictured left to right) answer your questions about anti-drug antibody and concentration assays for liposomal transgene enzymes
Was acid dissociation used in the ADA assay, and if not, how was the required drug tolerance achieved?
IJ: For most of the ADA assays, we tend to add an acid dissociation, mainly to improve drug tolerance. In this particular case, when we tried the use of drug tolerance, we observed that the signal baseline was significantly higher. The reason for this was that the assay was affecting the enzyme, causing aggregation and an increased baseline.
We noticed the heat was de-naturing the enzyme, as described in the PK assay. Therefore, we used heat to make the assay more transgene tolerant, which worked in this case.
What is the preferred approach to the transgene product PK assay?
IJ: The use of that surrogate heated matrix worked for us. The advantage of the heated matrix is that all the endogenous matrix is removed, and it keeps the matrix similar to the matrix that is deficient in the desired transgene product.
This leads to improved assay sensitivity, and the other matrix interference can be resolved at the same time. You do not want to get into sample analysis and realize you have poor parallelism. You must remember the transgene product is not what we are using as a calibrator. We want to ensure that the matrix interference you see in development is similar to that you see in the sample analysis.
You mentioned that the method for transgene quantification assessment can only be classified as fit for purpose after sample analysis. Can you elaborate on that?
IJ: This comes back to the use of reference material. For the PK assay, we have to use a well-characterized reference material and have the right paperwork.
In this case, we are using products that mimic expected transgene products. The quantification and validation results are not what we expected for the transgene product. We can claim the assay is fit for purpose once we get the samples that contain the actual transgene, quantify them, and carry out the parallelism to ensure the condition of these samples mirrors that of the surrogate product that we have used for validation.
When developing the transgene quantification assay you saw slightly higher sensitivity, better intra-assay precision, and parallelism with the Gyrolab platform compared to the MSD platform. Have you experienced these differences in results between the technologies for other types of assays?
IJ: With the PK assays that we develop internally, we adopt the principle of always trying to develop these assays on multiple platforms, so we can make better decisions.
In this case, the Gyrolab was better than the MSD. One of the biggest advantages of the Gyrolab platform is you can develop your assay quickly compared to the use of the MSD.
In some cases, MSD can give you the required sensitivity, but in this case, even though the MSD may have provided better sensitivity, the method had poor parallelism. The Gyrolab provided acceptable data in terms of parallelism, which was key in terms of what we were looking for here.
Why was the homogenous assay format beneficial for PK analysis when compared to the stepwise assay format?
IJ: For any form of PK, we usually prefer to use a stepwise format. The stepwise format is key in terms of avoiding things like the hook effect. With PK, you can have a very wide dynamic range. You do not want to underestimate the concentration of your drug in the sample.
In this case, we knew that the dynamic range required was narrow based on the context of use. Homogenous assays are specific, and in some cases also more sensitive. In this case, it was proven to be the most sensitive method, and the parallelism on the homogenous format was much better. As a result, we chose the homogenous format.
Are there any available guidelines on which type of CD to use depending on the assay?
JC: It depends on what the assay sensitivity requirements are. Most of the time, we recommend that assay development starts on either the 200 or the 1000 nanoliter CD. This ensures that you can screen reagents and test the assay range. If you need a very sensitive assay we would recommend you use the 4000 nanoliter CD.
If you want to measure very high concentrations, you may want to look at a 200-nanolitre CD. The advantage of the 200-nanolitre CD is it has 112 additional structures. This allows you to screen more reagents.
For the ADA assay, what was conjugated?
IJ: The same problem is present for either a PK assay or an ADA assay – you must find an alternative material that mimics your transgene product, and then assume that your transgene product will behave similarly. My best advice would be to get the exact transgene product that you want, by getting the transgene elsewhere. That would be the perfect case scenario for PK.
What percent of casein worked for the ADA?
IJ: We used 1% casein. It is worth bearing in mind that casein at 1% is still not very soluble. 1% gave us the desired baseline, but we had to implement additional strategies such as increased centrifugation and additional mixing step to be able to achieve the desired precision in the assay.
Biographies
John Chappell has approximately 25 years of experience in the Contract Research industry supporting both preclinical and clinical drug development. He has specialized in supporting biological compounds from an analytical perspective e.g. Pharmacokinetic, Immunogenicity and Biomarker analysis. He is particularly interested in validation requirements and ensuring that data generated will be acceptable to the regulatory authorities. He has spoken at many international conferences on various topics including Oligonucleotide analysis, Biomarker Analysis, Immunogenicity and the analytical support of Biosimilar programs. He now leads the Application Support and Service teams for Gyros Protein Technologies where he is responsible for customer service and technical support in Europe and the Asia Pacific regions. John has been a user of the Gyrolab® system for over 10 years so will use this experience to help customers. He is a Fellow of the Royal Society of Chemistry and was involved in the American Association of Pharmaceutical Scientists (AAPS) Biosimilar Committee that has prepared papers on Pharmacokinetic and anti-drug antibody assays.
Issa Jyamubandi received a BSc in Biomedical Sciences from the University of Coventry and a PhD from the University of East Anglia (UEA) developing targeted peptide drug conjugate therapies (PDCs) for melanoma. Issa has over 10 years’ experience developing and validating anti-drug antibodies (ADA), neutralizing antibodies (Nabs) and PK assay using various platform including the MSD, Gyrolab and various absorbance, fluorescence and luminescence platform. Issa is subject matter expert in the development and validation of ADA and Nabs assays for challenging therapeutic products ranging from small peptides (<5kDa), bispecific, ADCs, enzymes, Pegylated therapies, AAV gene therapy and transgene products.
Affiliations
John Chappell
Director of Scientific Support,
EMEA and Asia Pacific,
Gyros Protein Technologies
Issa Jyamubandi
Principal Scientist,
Alliance Pharma

Authorship & conflict of interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The authors have no conflicts of interests.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2023 Gyros Protein Technologies. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article is a transcript of a webinar, which can be found here.
Webinar recorded: Jan 26 2023; Revised manuscript received: Mar 7 2023; Publication date: Apr 4 2023