
Regulatory pathway on the manufacture & quality control of recombinant adeno-associated virus vectors
Cell Gene Therapy Insights 2018; 4(2), 89-99.
10.18609/cgti.2018.012
Human gene therapy vectors are rapidly emerging as a new class of licensed products for single gene disorders. Examples include a retroviral gene therapy of hematopoietic stem cells for severe combined immune deficiency due to adenosine deaminase deficiency (ADA-SCID), marketed as Strimvelis® in Europe, and a recombinant adeno-associated virus (rAAV) vector for inherited retinal dystrophy due to RPE65 deficiency (IRD), approved as Luxturna® in the USA. The regulatory regime surrounding the human use of investigational human gene therapy products in the USA and Europe has been developed in the context of multiple trials of these vectors in patients over the past 28 years. Aspects of this framework relevant for rAAV vectors are discussed here.
Overview of US Regulatory & Advisory Processes for rAAV Gene Therapy
Much of the regulatory process is identical to that for other biologics, but there are superimposed issues with gene therapy, such as immune responses to vector components and transgene products, genotoxicity (insertional mutagenesis) issues, vector biodistribution issues, and the use of molecular indicators in lieu of traditional pharmacokinetic and pharmacodynamic outcomes. The majority of US rAAV-gene therapy vector trials have been performed in academic medical centers. Such medical centers, as recipients of NIH funding are subject to processes dictated by the US Department of Health and Human Services (HHS) Protection of Human Subjects as outlined in the Code of Federal Regulations (45 CFR § 46 and 21 CFR § 50 ) [1]. This document provides the procedural requirements of investigators and institutions with particular focus on assuring compliance through the use of Institutional Review Boards (IRB) (21 CFR 56). The key elements of the review process for human gene therapy protocols at US academic medical centers are presented in Table 1, which include the Institutional Biosafety Committee (IBC), Office of Biotechnology Activities (OBA)/ Recombinant Advisory Committee (RAC), and US Food and Drug Administration (FDA) Center for Biologics Evaluation and Research (CBER).
| Table 1: Oversight and regulatory bodies responsible for rAAV gene therapy clinical trials. | |||
|---|---|---|---|
| Committee or group | Primary responsibility | Local vs national | Notes |
| Institutional Biosafety Committee (IBC) | Biosafety | Local | Most in-depth review for new vector classes |
| Office of Biotechnology Activities (OBA)/Recombinant Advisory Committee (RAC) | Biosafety, human subjects, policy | National | Usually email review unless there are novel biosafety or ethical issues |
| Institutional Review Board (IRB) | Human subjects protection | Local | Has primary role in reviewing informed consent, disclosures and conflicts |
| US Food and Drug Administration (FDA)-Center for Biologics Evaluation and Research (CBER) | Safety and efficacy of therapeutics, including investigational therapeutics | National | Reviews all aspects of preclinical, manufacturing and clinical trial |
| Funding Agency | At agency discretion | Various | Certain NIH institutes or Foundations may establish independent data monitors or DSMBs |
In practice, investigators initiating rAAV gene therapy trials must submit their proposed clinical trials, along with supporting information on proof-of-concept studies, and pre-clinical (non-clinical) safety studies, to the local IBC for review of the biosafety aspects of the work, and to the local IRB to review all aspects of the research relevant to human subject’s protection. The latter includes a basic presentation of the nature and purpose of the research, the subject selection (inclusion and exclusion) criteria, the potential risks and benefits, and the proposed outcome measures. Once the protocol has been approved by the local IBC (with any required modifications), the protocol may be forwarded to the NIH OBA and its RAC, along with specific items within the Appendix M, which address recombinant genetic products specifically [2,3]. Historically, the RAC was developed to provide a public forum for discussion of issues specific to gene therapy, but it is not truly a regulatory process. Thus, the particular role of the RAC has appropriately evolved over the years. Currently, the protocol-specific review role has been reduced, and the public meetings primarily deal with more global issues such as review for cases with truly novel biosafety or ethical implications [3,4]. Usually after this local and national review, CBER and its Office of Cellular, Tissue, and Gene Therapy (OCTGT) of the FDA oversees the actual human use of the rAAV vector.
US FDA Standards & Oversight
The US FDA must be notified of any intended use of investigational gene therapy vectors, by way of an investigational new drug (IND) application. The FDA has issued guidelines for industry for considerations when designing clinical trial for cell and gene therapy products [5]. The individual or entity submitting the IND application is termed the IND sponsor. The IND sponsor can be the study investigator, but often these functions are separated, with the entity possessing commercialization rights to the gene therapy vector most often serving as the IND sponsor. A sponsor may request a pre-IND meeting or teleconference with FDA-CBER prior to submission in order to ask specific questions that can facilitate the inclusion of all necessary data and materials and the rapid processing of the IND. As of May 5 2018, Commercial INDs and Master Files must be submitted using the electronic eCTD format; however, noncommercial INDs such as investigator-sponsored or expanded-access INDS are only encouraged to submit using the eCTD format [6].
At times, it is desirable to confer with the FDA at an even earlier stage. For example, investigators who have completed proof-of-concept studies in disease animal models will usually do these studies with research-grade vector and under standard laboratory conditions, rather than under formal FDA-Good Laboratory Practice (GLP) conditions. It is often useful to include in the IND a set of formal GLP or GLP-like pharmacology and toxicology (pharm-tox) studies with rAAV vector manufactured with the exact process to be used for the human trial. Such formal preclinical studies may be complex and costly. Therefore, it may be prudent to engage the FDA in a so-called ‘pre-pre-IND’ meeting or teleconference in which the team will present the basic outline of the proposed trial and the specific plan for the proposed GLP pharm-tox studies prior to initiating such studies. After the formal GLP studies are complete, the report from such studies can be included in the pre-IND discussion document, and then pre-IND conference can serve as a final check before the formal submission. However, in an industry setting, pre-pre IND meetings are more uncommon and generally are informal discussions. In the industry setting, the pre-IND meeting allows the agency to have input in the design of pharm-tox studies and how the vector will be evaluated, and therefore are performed before the starting the IND-enabling studies and pharm-tox. A typical sequence of studies and meetings leading to an IND application is shown in Figure 1.
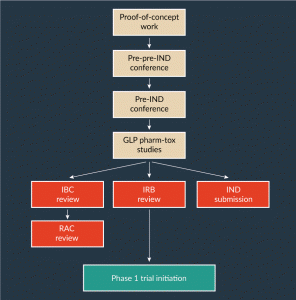
Figure 1: Example of a typical sequence of studies and regulatory approvals for an rAAV gene therapy product development plan.
GLP: Good Laboratory Practice; IBC: Institutional Biosafety Committee; IND: Investigational New Drug; IRB: Institutional Review Board; RAC: NIH Recombinant DNA Advisory Committee.
A detailed discussion of the design preclinical pharm-tox studies for gene therapy is beyond the scope of this review, but has been reviewed elsewhere [7]. Pharm-tox studies are usually conducted in two species, often a rodent and a larger animal; and while for other drugs and biological agents, one would ideally like to have pharm-tox safety data at a ten-fold higher dose than that used in humans, the limitations on rAAV manufacturing (and the maximal concentrations at which the vector may be stable in solution) may make it less feasible to do so. This approach has recently been called into question by data showing unanticipated toxicities in large animal models with very high doses of some AAV9-like vectors [8–10]. Also, the number of tissues to be analyzed for various assays of vector biodistribution and expression creates an unusual burden for laboratory analysis, but are conducted nonetheless [11]. For instance, a study involving three doses, three time-points, an n of 5 per dose/per sex (total 10), sets up a 90-animal study. If 14 organs are analyzed in triplicate by PCR, this would amount to 3,780 PCR reactions. One point worthy of note is that aspects of rAAV vector biodistribution are attributable to the specific capsid variant, dose and route and should not need to be repeated in detail if only the transgene differs from previously published results [12]; however, commercial entities may not be willing to rely on such data [12].
The basic components of the IND are indicated in Table 2. The most substantive sections are: the preclinical data set (reflecting the study outcomes from the proof-of-concept and GLP pharm/tox studies), the clinical protocol, and the chemistry, manufacturing and control (CMC) section. The clinical protocol proposed in the IND should be consistent with that presented to the local IRB, IBC and OBA-RAC. Ideally, rAAV clinical gene therapy protocols are performed under Good Clinical Practice (GCP) conditions. Such conditions include documented standard operating procedures (SOPs) for specific clinical procedures, which are reflected in an investigator study manual, a standardized system for capture of all study outcomes on case report forms (CRFs), and a system for reconciliation between primary source documents and CRFs.
| Table 2: Components of the IND application (specified in 21 CFR § 312) | |
|---|---|
| IND Section | Description |
| Form FDA 1571 (21 CFR 321.23(a)(1)) | Identifies product, sponsor, other key facts |
| Table of Contents (21 CFR 312.23(a)(2) | |
| Introductory Statement (21 CFR 312.23(a)(3) | Basic information on the product |
| General Investigational Plan (21 CFR 312.23(a)(3)) | Overview summary of research |
| Investigator’s Brochure (21 CFR 312.23(a)(5)) | Forms the basis for the future product label with indication, dose, risk and benefit |
| Protocol(s) (21 CFR 312.23(a)(6)) | Clinical trial outline and information on investigators and facilities |
| Study protocols (21 CFR312.23(a)(6)) | |
| Investigator Data (21 CFR 312.23(a)(6)(iii)(b)) or completed Form(s) FDA1572 | |
| Facilities data (21 CFR 312.23(a)(6)(iii)(b)) or completed Form(s) FDA1572 | |
| Institutional Review Board data (21 CFR 312.23(a)(6)(iii)(b)) or completed Form(s) FDA1572 | |
| Chemistry, Manufacturing and Control Data (21 CFR 312.23(a)(7)) Environmental assessment or claim of exclusion (21 CFR 312.23(a)(7)(iv)(e)) | Defines product, manufacturing and purification methods |
| Pharmacology and Toxicology Data (21 CFR 312.23(a)(8)) | Preclinical (animal) data on bioactivity, safety, biodistribution, etc. |
| Previous Human Experience (21 CFR 312.23(a)(9)) | May relate to vector or vector class |
| Additional Information (21 CFR 312.23(a)(10)) | |
| Biosimilar user Fee Cover Sheet (Form FDA 3792) | |
| Clinical Trials Certification of Compliance (Form FDA 3674) | |
The CMC section describes the rAAV gene therapy product itself, generally including a complete DNA sequence, details of the processes used for ‘upstream’ manufacturing and ‘downstream’ purification, and the array of quality control (QC) assays that have been performed on the product. In general, products for human use should comply with FDA standards around current Good Manufacturing Practice (cGMP). In the case of rAAV, the basic elements of the upstream processes that have been published to date are presented in Table 3, and have been reviewed elsewhere [13]. There are four specific elements that are required for any upstream rAAV process: a packaging cell; a source of AAV Rep (serotype 2) and Cap (capsid variant of choice) proteins; a source of adenovirus helper genes (E1a, E1b, E2a, E4 and VA-RNA), HSV1 or baculovirus helper function; and an AAV2-inverted terminal repeat (ITR)-flanked proviral vector of a size appropriate for packaging (less than 4.7kb). These four elements are presented in a variety of forms in the different packaging methods. Downstream purification has been described using a number of different methods as well, including CsCl and iodixanol ultracentrifugation [14,15], ion exchange chromatography [16,17], affinity chromatography [18–20] and tangential flow ultrafiltration [21–23].
| Table 3: Upstream rAAV packaging methods. | |||||
|---|---|---|---|---|---|
| Packaging method | Host cell | Source of helper genes | Source of Rep and Cap | Source of ITR-construct | Ref. |
| Triple transfection | HEK-293 | Helper plasmid and host cell | Second helper plasmid | Proviral plasmid | [31] |
| Double transfection | HEK-293 | Dual helper plasmid and host cell | Dual helper plasmid | Proviral plasmid | [32,33] |
| Baculovirus | Sf-9 | rBEV-Rep/cap | rBEV-Rep/cap | rBEV-rAAV | [20,34] |
| HSV1 helper | Vero-27, BHK, HEK-293 | rHSV1 | rHSV1-rep/cap | rHSV1-rAAV | [35–38] |
| Rescuable HeLa lines | HeLa | rcAd5 or rcAd2 | Integrated in HeLa | Integrated in HeLa | [39] |
The CMC section will define the source and quality control of all constituents of the manufacturing process and QC assays performed on constituents, intermediate components of the product, and the final product itself. The final product will be tested by a number of different categories of assays, as shown in Table 4. The final product will be assayed for general safety, such as sterility, absence of endotoxin and mycoplasma. Others assays will determine residual components of the manufacturing system, such as cellular DNA, plasmid DNA or helper virus DNA. Components of the packaging system can theoretically generate replication competent (rc) versions of AAV or the helper virus, and these should be assayed for. Assessment of impurities of clinical-grade AAV products have been described and reviewed elsewhere [24,25]. Finally, there must be measures of the identity, activity and purity of the final product after purification. Some of these QC assays define the quality sufficient for release of the lot of vector and those that may simply be reported without defining a critical value. In addition to assays on the final product reported as part of the lot release, additional stability testing of the product may be performed over time. Stability testing is generally limited to essential test of vector titer and biologic activity. The duration of stability testing is usually designed to match the intended duration of storage prior to administration to the final cohort of subjects.
| Table 4: Types of QC assays typically performed on the final product in rAAV gene therapy preparations. | |
|---|---|
| Assay type | Examples of specific tests |
| General safety | Sterility, mycoplasma,adventitious, endotoxins |
| Contaminants specific to the process | Plasmid DNA, Cell line DNA, rc-AAV, rc-Ad, rc-HSV |
| rAAV identity, titer and purity | Vector genome sequence, vector genome titer, physical titer, infectious titer, presence of other proteins |
| Transgene activity | Transgene expression in cell line or other after AAV transduction |
Ongoing Regulatory Oversight
Once the IND is submitted, FDA will respond to the sponsor within 30 days if there is an issue indicating a clinical hold contingent on providing additional information or modification. Subsequent to initiation of the trial, ongoing trial reporting is required to those entities initially involved in the review. Serious adverse events (SAEs) must be reported, usually within 48 hours to IRB and FDA, while other adverse events (AEs) are reported at predetermined intervals. Data relevant to safety of enrollment of future patients in human gene therapy trials must also be reported to IBC and OBA-RAC. Even in the absence of SAEs, protocols that include a dose-escalation often require interim review of safety data (and perhaps bioactivity data) with regulatory authorities and/or DSMB review between dosage cohorts or even between individual patients as they are enrolled. The manufacturing process will adhere to cGMP principles throughout, but the rigor of cGMP compliance of specific practices often progressively increases from Phase 1 material through commercial licensure. For example, the frequency and tolerance levels for facility monitoring required may be relatively loose in a facility used to manufacture the Phase 1 material, but extremely rigorous in a commercial cGMP facility.
Regulatory Oversight of rAAV Trials in Europe
The European Union (EU) uses the term Advanced Therapy Medicinal Products (ATMPs) to describe gene therapy vectors, tissue engineered products and somatic cell therapies. The European Medicine Agency (EMA) has issued guidelines on clinical aspects of gene therapy products [26,27]. It is up to individual member states to approve clinical trials, but the EMA is involved in marketing and licensing products. However, major changes to clinical trial regulation will occur in 2019 to harmonize the assessment and processes of clinical trials throughout the EU. The common regulatory framework for ATMPs was described in regulations adopted by the EU parliament in 2007 [28]. Specifically, rAAV vectors are reviewed by the Committee on Advanced Therapeutics (CAT), after which they are reviewed by the EMA Committee for Medicinal Products for Human Use (CHMP), which issues an opinion. The final step is formal approval from the European Commission. Attempts to harmonize requirements between FDA, EMA and Japanese regulators have moved forward through the International Committee on Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH guidelines provide consistency of the format for submission of materials analogous to those in the Appendix M and the IND. This does not imply that the judgment of the relative risk of various aspects of products will be viewed identically. The EU process regarding cGMP compliance is similar to that of the FDA, although each agency may emphasize different aspects. For instance, the absolute restriction on bovine serum in cGMP processes in Europe represents a greater level of perceived risk from transmissible spongiform encephalopathies (TSE). There may be other specific points of emphasis depending on the process. In general, however, the ICH regime has provided a very useful framework for achieving approvals for commercial use of gene therapies across multiple countries of North America, Europe and Japan.
Additional Special Considerations & Best Practices
The fact that rAAV gene therapy vectors are being studied and approved for treatment of severe, life-threatening or vision-threatening genetic conditions has raised a number other complex issues. First of all, many of the rAAV gene therapy trials are being performed in infants and children. In the USA, infants and children have been identified as a special vulnerable population. Studies in infants and children are held to a higher standard than those done in adults relative to the risk-benefit balance. Specifically, only ‘minimal risk’ procedures are allowed in children if there is no prospect for benefit from a particular study (Subpart D, 45 CFR § 46.401) [1,29]. If the potential for benefit is only indirect, i.e., by providing knowledge for future treatment of their disease, then only a ‘minor increase’ above minimal risk is allowed in children. Minimal risk is clearly defined as the level of risk associated with normal activities of living, such as riding in an automobile or bathing. Minor increase above minimal risk is equivalent to the level of risk associated with the standard treatment for their underlying condition. Higher levels of risk are allowable in adults, who give informed consent for themselves.
Another issue specific to life-threatening conditions, particularly those affecting infants and children is the risk of ‘therapeutic misconception’, i.e., the misunderstanding by the patient that their own individual health status is likely to be improved by participating in a gene therapy trial, even in instances where this is very unlikely to be the case. Therapeutic misconception has commonly been encountered in human gene therapy trials, in spite of efforts by investigators and sponsors to be very explicit in forms designed to provide informed consent. One tool that has been used to identify and avoid therapeutic misconception is a practice known as ‘decision monitoring’ in which an independent individual contacts the research volunteer after they have given consent and explores their knowledge of their likelihood of health benefit from participation.
The potential for financial conflicts of interest is also a topic of special interest in this field. Many pioneering gene therapy investigators have also been inventors of technology on which rAAV gene therapies are based. Justifiably, many such investigators have a strong desire to participate in the first-in-human studies of their vectors, and they may indeed possess insights into the diseases and the vectors that will benefit the participants. The best practices to protect human subjects from investigator bias in such circumstances may include: limitations on the equity interest of investigators in firms commercializing gene therapy; full disclosure of potential conflicts in informed consent documents, publications and presentations; and independent oversight of study conduct where possible. In a related issue, the extremely high cost per dose of rAAV gene therapy has presented concerns about both the financial sustainability of such therapies and the biomedical ethics of such expenditures. Interestingly, rAAV gene therapies are not alone in this excessive cost. A recent analysis indicated that modern, molecular-based therapies of many different types (e.g., targeted small molecules, monoclonal antibodies, enzyme replacement therapies) present costs ranging from $250,000 to $500,000 per year [30] and such returns have created a robust incentive for investment in developing breakthrough therapies for rare genetic disorders.
Future of rAAV Gene Therapy & Its Regulation
The future of rAAV-based gene therapies for genetic diseases seems particularly bright. This is even more poignant given the long history of medicine viewing genetic diseases as intrinsically incurable. The chances seem particularly good for current rAAV platforms to treat numerous genetic conditions of the retinal, muscle, liver and central nervous system, since the target cells in these tissues are generally non-dividing and accessible to transduction with known rAAV serotypes. The current regulatory schema would seem to fit each of these applications well, with the primary challenge being scaling up manufacturing sufficiently to treat substantial numbers of patients, especially in the case of target organs of larger mass, such as liver and muscle. Future applications of rAAV-based delivery of CRISPR/Cas9 or other genome editing tools may require some modification of the existing regulatory approach, in order to understand greater risks of off-target genotoxicity or germ line alteration. These and even more targeted molecular therapies promise to further revolutionize modern medicine through the 21st century and beyond.
Financial & competing interests disclosure
TRF is a paid scientific advisor for Beam Therapeutics. AMK and TRF are inventors on a pending patent application related to technology described in this paper. No writing assistance was utilized in the production of this manuscript.
References
1. Protections, O.f.H.R., 45, U.S.D.o.H.a.H. Services, Editor.
2. Policy, O.o.S., NIH Guidelines for Research involving recombinant or synthetic nucleic acid molecules, D.o.H.a.H. Services, Editor 2016.
3. Oversight and Review of Clinical Gene Transfer Protocols: Assessing the Role of the Recombinant DNA Advisory Committee, Lenzi RN, Altevogt BM, and Gostin LO (Eds). DC, USA (2014).
4. Rainsbury, JM. Biotechnology on the RAC–FDA/NIH regulation of human gene therapy. Food Drug Law J. 2000; 55(4): 575–600.
5. Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products: Website
6. Electronic Common Technical Document: Website
7. Flotte TR, Conlon TJ, Mueller C. Preclinical study design for rAAV. Methods Mol. Biol. 2011; 807: 317–37.
CrossRef
8. Flotte TR, Buning H. Severe Toxicity in Nonhuman Primates and Piglets with Systemic High-Dose Administration of Adeno-Associated Virus Serotype 9-Like Vectors: Putting Patients First. Hum. Gene Ther. 2018.
CrossRef
9. Hordeaux J et al. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018.
https://doi.org/10.1016/j.ymthe.2018.01.018″ rel=”noopener” target=”_blank”>CrossRef
10. Hinderer C et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018.
CrossRef
11. Flotte TR, Conlon TJ, Poirier A, Campbell-Thompson M, Byrne BJ. Preclinical characterization of a recombinant adeno-associated virus type 1-pseudotyped vector demonstrates dose-dependent injection site inflammation and dissemination of vector genomes to distant sites. Hum. Gene Ther. 2007; 18(3): 245–56.
CrossRef
12. Kiem HP, Baum C, Bushman FD et al. Charting a clear path: the ASGCT Standardized Pathways Conference. Mol. Ther. 2014; 22(7): 1235–8.
CrossRef
13. Robert MA, Chahal PS, Audy A, Kamen A, Gilbert R, Gaillet B. Manufacturing of recombinant adeno-associated viruses using mammalian expression platforms. Biotechnol. J. 2017; 12(3).
CrossRef
14. Strobel B, Miller FD, Rist W, Lamla T. Comparative Analysis of Cesium Chloride- and Iodixanol-Based Purification of Recombinant Adeno-Associated Viral Vectors for Preclinical Applications. Hum. Gene Ther. Methods 2015; 26(4): 147–57.
CrossRef
15. Zolotukhin S, Byrne BJ, Mason E. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999; 6(6): 973–85.
CrossRef
16. Kaludov N, Handelman B, Chiorini JA. Scalable purification of adeno-associated virus type 2, 4, or 5 using ion-exchange chromatography. Hum. Gene Ther. 2002; 13(10): 1235–43.
CrossRef
17. Smith RH, Yang L, Kotin RM. Chromatography-based purification of adeno-associated virus. Methods Mol. Biol. 2008; 434: 37–54.
CrossRef
18. Qiu J, Handa A, Kirby M, Brown KE. The interaction of heparin sulfate and adeno-associated virus 2. Virology 2000; 269(1): 137–47.
CrossRef
19. Auricchio A, O’Connor E, Hildinger M, Wilson JM. A single-step affinity column for purification of serotype-5 based adeno-associated viral vectors. Mol. Ther. 2001; 4(4): 372–4.
CrossRef
20. Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 2009; 17(11): 1888–96.
CrossRef
21. Lock M, Alvira M, Vandenberghe LH et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther. 2010; 21(10): 1259–71.
CrossRef
22. Doria M, Ferrara A, Auricchio A. AAV2/8 vectors purified from culture medium with a simple and rapid protocol transduce murine liver, muscle, and retina efficiently. Hum. Gene Ther. Methods 2013; 24(6): 392–8.
CrossRef
23. Qu W, Wang M, Wu Y, Xu R. Scalable downstream strategies for purification of recombinant adeno- associated virus vectors in light of the properties. Curr. Pharm. Biotechnol. 2015; 16(8): 684–95.
CrossRef
24. Wright JF. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines 2014; 2(1): 80–97.
CrossRef
25. Reflection paper on quality, non-clinical and clinical issues related to the development of recombinant adeno-associated viral vectors: Website
26. Guideline on the quality, non-clinical and clinical aspects 5 of gene therapy medicinal products: Website
27. Clement N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016; 3: 16002.
CrossRef
28. REGULATION (EC) No 1394/2007 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004, O.J.o.t.E. Union, Editor. 2007.
29. Flotte TR, Frentzen B, Humphries MR, Rosenbloom AL. Recent developments in the protection of pediatric research subjects. J. Pediatr. 2006; 149(3): 285–6.
<a href=”https://doi.org/10.1016/j.jpeds.2006.04.033
30. Flotte TR. Ethical Implications of the Cost of Molecularly Targeted Therapies. Hum. Gene Ther. 2015; 26(9): 573–4.
CrossRef
31. Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998; 72(3): 2224–32.
32. Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 1998; 9(18): 2745–60.
CrossRef
33. Collaco RF, Cao X, Trempe JP. A helper virus-free packaging system for recombinant adeno-associated virus vectors. Gene 1999; 238(2): 397–405.
CrossRef
34. Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002; 13(16): 1935–43.
CrossRef
35. Conway JE, Zolotukhin S, Muzyczka N, Hayward GS, Byrne BJ. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J. Virol. 1997; 71(11): 8780–9.
36. Feudner E, de Alwis M, Thrasher AJ, Ali RR, Fauser S. Optimization of recombinant adeno-associated virus production using an herpes simplex virus amplicon system. J. Virol. Methods 2001; 96(2): 97–105.
CrossRef
37. Conway JE, Rhys CM, Zolotukhin I et al. High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene Ther. 1999; 6(6): 986–93.
CrossRef
38. Thomas DL, Wang L, Niamke J et al. Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum. Gene Ther. 2009; 20(8): 861–70.
CrossRef
39. Martin J, Frederick A, Luo Y et al. Generation and characterization of adeno-associated virus producer cell lines for research and preclinical vector production. Hum. Gene Ther. Methods 2013; 24(4): 253–69.
CrossRef
Affiliations
Terence R Flotte & Allison M Keeler-Klunk
Horae Gene Therapy Center and Department of Pediatrics, University of Massachusetts Medical School, MA, USA

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License</