Progressing towards a cure for deafness through gene therapy
Cell Gene Therapy Insights 2016;2(2), 175-182
10.18609/cgti.2016.020
Submitted: Apr 15 2016 Published: Jul 5 2016
Hearing loss
Acquired and hereditary deafness affect an estimated 360 million people worldwide (World Health Organization, 2013). There are currently no pharmacological interventions on the market for hearing loss despite recent advances in our understandings of the capacity of the cochlea for repair and regeneration. However, gene therapy is emerging as a promising method to deliver factors necessary for recovery of hearing. In the cochlea, there are some challenges to overcome before gene therapy can be considered in the clinic, including efficiency and specificity of transduction and overcoming the cellular degenerative changes that accompany hearing loss.
Gene therapy in the cochlea
The cochlea houses the organ of Corti: the hearing sensory organ that contains one row of inner hair cells, three rows of outer hair cells, supporting cells and innervating auditory nerve fibers from spiral ganglion neurons (Figure 1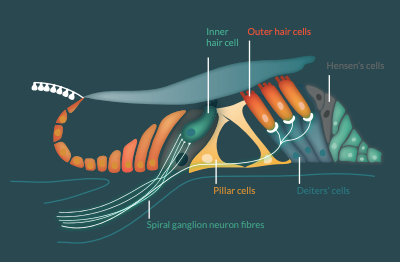
Direct injection of viral vectors (e.g., adenovirus or adeno-associated virus; [AAV]) into cochlear fluids through a small hole drilled into the cochlear wall (cochleostomy) or through the round window membrane of the cochlea ensures localised gene transduction, but with extremely variable efficiency. For example, the number of transduced hair cells can be as low as 15% or as high as 100% following injection with AAV (serotype 1 or 8) into mouse cochleae, with age being a contributing factor [2,3]. A sealed injection system is also critical, as the fluids of the cochlea readily leak out during injection and severely reduce the transduction efficiency [4]. Unfortunately, loss of residual hearing can occur with direct injection into the cochlear fluids [5,6]. To overcome this hearing loss and to maintain a sealed cochlea, the permeability of the round window membrane to some viral vectors has been exploited, with factors such as hyaluronic acid shown to enhance its permeability to adenovirus and Sendai virus [7-9]. The cochlear implant has also been used as a delivery device for gene therapy. A DNA gene construct was introduced to the cochlea via a modified cochlear implant where electrical stimulation from the implant itself was used to electroporate cells in the cochlea, with resulting gene expression in cells lining the scala tympani [10].
Cell specificity of gene expression is also important and this can be achieved in the cochlea in a number of ways: utilizing the anatomy of the cochlea (injection into different fluid chambers of the cochlea [5,11-13]); different serotypes of AAV, which naturally target different cell types of the cochlea; and specific targeting of hair cells or neurons with cell-specific promoters [14-16].
Specific targeting of hair cells or other cells of interest and safe and efficient injection techniques will be necessary to develop effective molecular therapies for functional hearing protection and/or recovery.
Gene therapy for acquired hearing loss
Two of the key cell types affected by acquired deafness are cochlear hair cells and neurons. Protecting and regenerating these cells via gene therapy are key steps towards reversing hearing loss.
Neurotrophin gene therapy
Neurotrophins are diffusible proteins, which are critical for neuronal fiber outgrowth and patterning during cochlear development and also for continued neural survival. The necessity to promote neural survival after deafness is of critical importance as neurons are the targets of electrical stimulation from cochlear implants.
Neurotrophin gene therapy in the cochlea has been shown to prevent neuronal degeneration after ototoxic-induced hearing loss for up to 6 months, with the effect localized to the region most proximal to the injection site where the highest level of viral transduction occurs [17-20]. In addition to protecting total neuron number, neurotrophin gene therapy also promoted localized and directional peripheral fibre resprouting, highlighting the potential to enhance the nerve-electrode interface of a cochlear implant or to direct the growth of fibers back to regenerated hair cells [5,17,21].
Recent findings strongly suggest neurotrophin gene therapy is a promising approach to enhance auditory nerve survival for an extended period [17,18,21]. However, if the neurotrophin gene therapy is targeted to the organ of Corti, neuronal survival is not sustained beyond 6 months in guinea pigs due to continued degenerative changes associated with acquired hearing loss. The time between hearing loss onset and treatment, and hence the level of degeneration, has also been shown to critically reduce the efficacy of neurotrophin gene therapy [22].
Neurotrophin gene therapy has been shown to be extremely effective in protecting auditory neurons and stimulating the regrowth of auditory nerve fibres. Targeting the therapy to cells that do not degenerate after hearing loss, such as cells lining the scala tympani or glial cells will result in continual release of neurotrophins to ensure long-term outcomes. However, more research is needed to link neural survival to improved cochlear implant function and whether this improvement is clinically meaningful.
Atonal gene therapy
Atoh1 is a transcription factor necessary for both hair cell development and survival and forced expression of Atoh1 in supporting cells of the organ of Corti initiates their trans-differentiation into ectopic hair cells [11,23-27].
The rates of conversion of hair cells and hearing recovery is highly dependent on age, degree of hearing loss and method of Atoh1 transduction [11,28,29]. In utero gene transfer of Atoh1 in mice results in ectopic, induced, functional hair cells, but when applied after birth Atoh1 is less effective, as demonstrated by poor hearing recovery outcomes and lack of mature markers in induced hair cells [11,28,29]. However, there have been some sporadic reports of partial recovery of hearing and balance function by Atoh1 in mature animals, suggesting that recovery of function is highly dependent on cellular context and level of pathology [27,30-35]. To improve therapy efficacy, it has also being suggested that regenerative approaches could consider reprogramming the senescent supporting cells post-deafness, via a small-molecule-based approach, enabling them to better respond to differentiating factors such as Atoh1 [36].
After sudden, severe hearing loss, there is only a short window of opportunity (at least in animal hearing loss models) during which residual supporting cells can be transformed into induced hair cells with Atoh1 gene therapy due to loss of differentiated supporting cells for transdifferentiation [11,27,37]. This window of opportunity is greater in a progressive, partial hair cell loss model, which may more accurately reflect human hearing loss pathologies [12]. Interestingly, it was shown that Atoh1 gene therapy after simulated gun-shot noise exposure could improve hearing thresholds through the repair of stereocilia of residual hair cells rather than the regeneration of new hair cells, showing a function of Atoh1 beyond de novo regeneration that could be developed into a molecular therapy for hearing recovery [38].
Developmentally, it is known that the expression pattern and level of Atoh1 is tightly and dynamically regulated [25,39,40]. Commonly used in Atoh1 gene therapy studies are adenoviral vectors with strong cytomegalovirus promoters for constitutive gene expression, which fail to mirror endogenous expression patterns and may impact on survival, maturation, and function of the resulting hair cells. Moreover, after transdifferentiation into induced hair cells, a continued but lower Atoh1 expression level may be essential for maintenance of induced hair cells via its regulation of expression of other genes known to be involved in hair cell survival such as Gfi1, Pou4f3, Neurog1 and Barhl1 [39,41,42]. The development of doxycycline-based conditional and inducible Atoh1 expression systems will prove useful in studying regulated Atoh1 expression in vivo, but is challenging with the blood–labyrinth barrier excluding doxycycline from the cochlea, preventing its clearance, and contributing to hearing loss [43-45].
Transforming supporting cells into hair cells will negatively impact on the cytoarchitecture and function of the organ of Corti, with supporting cells being critical for cochlear function. For recovery of function, it will be necessary to first promote the proliferation of supporting cells prior to Atoh1 gene therapy. Supporting cells from adult cochleae maintain the capacity to divide with therapeutic manipulation, with co-transfection of Pax2 and Atoh1 resulting in regenerated hair cells in vitro [46,47].
The prospect of regenerating hair cells after hearing loss is exciting and understanding the molecular dynamics involved in creating a mature functional hair cell from existing cells in the cochlea will help to develop a therapy that might require more sophisticated regulation of Atoh1 expression or more than one gene.
Gene therapy for hereditary deafness
At least half of all childhood deafness is inherited, with at least 80 deafness genes identified to date. Using mouse models of human hereditary deafness, a number of recent reports have shown significant progress towards hearing recovery following gene therapy.
In VGLUT3 knockout mice, there is a defect in the release of glutamate from inner hair cell afferent synapses. When AAV1–VGLUT3 was delivered to the round window membrane of the cochlea of mice at postnatal day 1–10, there were measurable improvements in hearing function within 7 days and complete restoration of function by 14 days, extending up to 9 months after gene delivery [2]. Connexin26 mutations account for a significant number of hereditary deafness cases and deletion of connexin26 in mice results in significant degeneration of the organ of Corti and auditory neurons. Injection of AAV–Cx26 into the scala media of Cx26 knockout mice at P0–1 improved morphology and cell communication in the organ of Corti but did not recover hearing, while degeneration of auditory neurons was reversed by neurotrophin gene therapy in 1 month old mice [48,49].
Many deafness syndromes are the result of mutations in hair cell stereocilia such as DFNB31 or type II Usher syndrome and are modelled by Whirler mice with affected stereocilia. Injection of AAV8–Whirlin through the round window membrane resulted in Whirlin expression in the tips of the stereocilia and restoration of stereocilia structure. Liekwise, the Tmc1 gene (affected in human DFNB7/11 and DFNA36) was introduced into mice with a targeted deletion of Tmc1 (Beethoven mice) via AAV2/1 (via the round window membrane of P0–2 mice), with resulting expression in the tips of stereocilia and recovery of hearing thresholds, although not to wild-type levels [50].
These studies emphasize the potential for gene therapy to counteract the functional defects of various underlying genetic faults that lead to loss of hearing. With further developments gene therapy could result in restoration of normal hearing function for many types of hereditary deafness with long-term or even life-long benefit.
Conclusions
The prevalence of hearing loss necessitates the development of a therapy that can prevent the degenerative changes associated with hearing loss and even restore hearing. Gene therapy is of particular interest for the correction of genetic defects that affect hearing and for the restoration of hair cells and neurons after acquired hearing loss.
For translation of potential gene therapies for hearing loss there is the need to optimise methods of gene transduction. This includes improving gene targeting to specific subpopulations of cells in the cochlea, controlling induced gene expression and optimising transfection efficiency through improved viral vectors and method of injection [1,51]. The round window membrane of the cochlea will be the most likely method of delivery of gene therapy in humans due to its accessibility and potential for hearing preservation, but overall transduction rates need to be increased for hearing recovery to be clinically meaningful [52-54]. This will necessitate the development of a gene therapy delivery system for the cochlea that prevents losses during injection and/or indirect methods that do not compromise the cochlea such as diffusion across the round window membrane.
A major challenge to overcome is the rapid decline in the efficiency and effectiveness of gene therapy with age. Most gene therapy studies with reported hearing recovery are performed in neonatal and early post-natal mice, highlighting the need for early diagnosis and intervention for congenital deafness and more research on gene therapy in mature subjects; the main group in the population with acquired hearing loss.
In parallel to improving general gene therapy techniques, Atoh1 gene therapy in particular will benefit from a greater understanding of the timing and dosage of Atoh1 and the co-factors required for mature hair cell formation, both developmentally and after injury to the cochlea [35]. A one-gene approach may have some role, perhaps in hair cell repair, but the best chance of recovering hearing may come from co-expression of Atoh1 with other factors that will initially help supporting cells to divide but also other factors such as Gfi1, Pou4f3, Neurog1 and Barhl1 that will help induced hair cells to fully differentiate, attract nerve fibers and survive long-term.
Collectively, these studies demonstrate that gene therapy holds promise for curing deafness, however, the efficacy of cochlear gene therapy in a human patient population remains to be determined. Medical researchers are currently conducting a clinical trial to assess the safety and efficacy of Atoh1 therapy in those suffering severe acquired hearing loss. The findings of this study will provide useful insight into the overall safety and efficacy of viral-mediated gene therapy. However, critical factors such as expression levels and duration, degree of pathology and age outlined above go beyond the scope of this initial trial and will also need to be addressed to more fully elucidate the efficacy of Atoh1 gene therapy in the desired patient population. There is much to be hopeful for and a deeper understanding of the molecular nature of hair cell formation and function, along with engineering advances in viral delivery techniques, will lead to great advances in the near future.
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
Acknowledgements
The authors of this manuscript are supported by the National Health and Medical Research Council GNT1024350, the Child Health Research Institute and Lucile Packard Foundation for Children’s Health, Stanford CTSA (UL1 TR001085). The Bionics Institute acknowledges the support it receives from the Victorian Government through its Operational Infrastructure Support Program.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1. Richardson RT, Atkinson PJ. Atoh1 gene therapy in the cochlea for hair cell regeneration. Expert Opin Biol. Ther. 2015;15, 417–30. CrossRef
2. Akil O, Seal RP, Burke K et al. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 2012; 75, 283–93. CrossRef
3. Chien WW, Isgrig K, Roy S et al. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Mol. Ther. 2016; 24, 17–25. CrossRef
4. Salt AN, Sirjani DB, Hartsock JJ et al. Marker retention in the cochlea following injections through the round window membrane. Hear. Res. 2007; 232, 78–86. CrossRef
5. Wise AK, Hume CR, Flynn BO et al. Effects of localized neurotrophin gene expression on spiral ganglion neuron resprouting in the deafened cochlea. Mol. Ther. 2010; 18, 1111–22. CrossRef
6. Ye Q, Tillein J, Hartmann R et al. Application of a corticosteroid (Triamcinolon) protects inner ear function after surgical intervention. Ear Hear. 2007; 28, 361–9. CrossRef
7. Goycoolea MV. Clinical aspects of round window membrane permeability under normal and pathological conditions. Acta Otolaryngol. 2001; 121, 437–47. CrossRef
8. Shibata SB, Cortez SR, Wiler JA et al. Hyaluronic acid enhances gene delivery into the cochlea. Hum. Gene. Ther. 2012; 23, 302–10. CrossRef
9. Kurioka T, Mizutari K, Niwa K et al. Hyaluronic acid pretreatment for Sendai virus-mediated cochlear gene transfer. Gene Ther. 2016; 23, 187–95. CrossRef
10. Pinyon JL, Tadros SF, Froud KE et al. Close-field electroporation gene delivery using the cochlear implant electrode array enhances the bionic ear. Sci. Transl. Med. 2014; 6, 233ra54. CrossRef
11. Atkinson PJ, Wise AK, Flynn BO et al. Hair cell regeneration after ATOH1 gene therapy in the cochlea of profoundly deaf adult guinea pigs. PLoS One 2014; 9(7): e102077. CrossRef
12. Wise AK, Flynn BO, Atkinson PJ et al. Regeneration of cochlear hair cells with Atoh1 gene therapy after noise-induced hearing loss. J. Regen. Med. 2015; 4, 1. CrossRef
13. Shibata SB, Di Pasquale G, Cortez SR et al. Gene transfer using bovine adeno-associated virus in the guinea pig cochlea. Gene Ther. 2009; 16(8): 990–7. CrossRef
14. Liu Y, Okada T, Sheykholeslami K et al. Specific and efficient transduction of Cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol. Ther. 2005; 12, 725–33. CrossRef
15. Liu Y, Okada T, Nomoto T et al. Promoter effects of adeno-associated viral vector for transgene expression in the cochlea in vivo. Exp. Mol. Med. 2007; 39, 170–5. CrossRef
16. Luebke AE, Foster PK, Muller CD et al. Cochlear function and transgene expression in the guinea pig cochlea, using adenovirus- and adeno-associated virus-directed gene transfer. Hum. Gene Ther. 2001; 12, 773–81. CrossRef
17. Atkinson PJ, Wise AK, Flynn BO et al. Neurotrophin gene therapy for sustained neural preservation after deafness. PLoS One 2012; 7, e52338. CrossRef
18. Atkinson PJ, Wise AK, Flynn BO et al. Viability of long-term gene therapy in the cochlea. Sci. Rep. 2014; 4, 4733. CrossRef
19. Shibata SB, Cortez SR, Beyer LA et al. Transgenic BDNF induces nerve fiber regrowth into the auditory epithelium in deaf cochleae. Exp. Neurol. 2010; 223, 464–72. CrossRef
20. Staecker H, Gabaizadeh R, Federoff H et al. Brain-derived neurotrophic factor gene therapy prevents spiral ganglion degeneration after hair cell loss. Otolaryngol. Head Neck Surg. 1998; 119, 7–13. CrossRef
21. Budenz CL, Wong HT, Swiderski DL et al. Differential effects of AAV.BDNF and AAV.Ntf3 in the deafened adult guinea pig ear. Sci. Rep. 2015; 5, 8619. CrossRef
22. Wise AK, Tu T, Atkinson PJ et al. The effect of deafness duration on neurotrophin gene therapy for spiral ganglion neuron protection. Hear. Res. 2011; 278, 69–76. CrossRef
23. Bermingham N, Hassan B, Price S et al. Math 1: An essential gene for the generation of inner ear hair cells. Science 1999; 284, 1837–41. CrossRef
24. Woods C, Montcouquiol M, Kelley MW. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat. Neurosci. 2004; 7, 1310–8. CrossRef
25. Cai T, Seymour ML, Zhang H et al. Conditional deletion of Atoh1 reveals distinct critical periods for survival and function of hair cells in the organ of Corti. J. Neurosci. 2013; 33, 10110–22. CrossRef
26. Kawamoto K, Ishimoto S, Minoda R et al. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J. Neurosci. 2003; 23, 4395-–400. Website
27. Izumikawa M, Minoda R, Kawamoto K et al. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat. Med. 2005; 11, 271–6. CrossRef
28. Gubbels SP, Woessner DW, Mitchell JC et al. Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 2008; 455, 537–41. CrossRef
29. Liu ZY, Dearman JA, Cox BC et al. Age-Dependent In Vivo Conversion of Mouse Cochlear Pillar and Deiters’ Cells to Immature Hair Cells by Atoh1 Ectopic Expression. J. Neurosci. 2012; 32, 6600–10. CrossRef
30. Mizutari K, Fujioka M, Hosoya M et al. Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron 2013; 77, 58–69. CrossRef
31. Kraft S, Hsu C, Brough DE et al. Atoh1 induces auditory hair cell recovery in mice after ototoxic injury. The Laryngoscope 2013; 123, 992–9. CrossRef
32. Schlecker C, Praetorius M, Brough DE et al. Selective atonal gene delivery improves balance function in a mouse model of vestibular disease. Gene Ther. 2011; 18, 884–90. CrossRef
33. Staecker H, Praetorius M, Baker K et al. Vestibular hair cell regeneration and restoration of balance function induced by math1 gene transfer. Otol. Neurotol. 2007; 28, 223–31. CrossRef
34. Jahan I, Pan N, Fritzsch B. Opportunities and limits of the one gene approach: the ability of Atoh1 to differentiate and maintain hair cells depends on the molecular context. Front. Cell. Neurosci. 2015; 9, 26. CrossRef
35. Jahan I, Pan N, Kersigo J et al. Beyond generalized hair cells: molecular cues for hair cell types. Hear. Res. 2013; 297, 30–41. CrossRef
36. Waldhaus J, Cimerman J, Gohlke H et al. Stemness of the organ of Corti relates to the epigenetic status of Sox2 enhancers. PLoS One 2012; 7, e36066. CrossRef
37. Taylor RR, Jagger DJ, Forge A. Defining the cellular environment in the organ of Corti following extensive hair cell loss: a basis for future sensory cell replacement in the Cochlea. PLoS One 2012; 7, e30577. CrossRef
38. Yang SM, Chen W, Guo WW et al. Regeneration of stereocilia of hair cells by forced Atoh1 expression in the adult mammalian cochlea. PLoS One 2012; 7, e46355. CrossRef
39. Jahan I, Pan N, Kersigo J et al. Neurog1 can partially substitute for Atoh1 function in hair cell differentiation and maintenance during organ of Corti development. Development 2015; 142, 2810–21. CrossRef
40. Pan N, Jahan I, Kersigo J et al. A novel Atoh1 “self-terminating” mouse model reveals the necessity of proper Atoh1 level and duration for hair cell differentiation and viability. PLoS One 2012; 7, e30358. CrossRef
41. Xiang M, Gao WQ, Hasson T et al. Requirement for Brn-3c in maturation and survival, but not in fate determination of inner ear hair cells. Development 1998; 125, 3935–46. Website
42. Wallis D, Hamblen M, Zhou Y et al. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 2003; 130, 221–32. CrossRef
43. Parker MA, Cheng YF, Kinouchi H et al. An independent construct for conditional expression of atonal homolog-1. Hum. Gene Ther. Methods 2014; 25, 1–13. CrossRef
44. Walters BJ, Zuo J. A Sox10(rtTA/+) Mouse Line Allows for Inducible Gene Expression in the Auditory and Balance Organs of the Inner Ear. J. Assoc. Res. Otolaryngol. 2015; 16, 331–45. CrossRef
45. Cox BC, Dearman JA, Brancheck J et al. Generation of Atoh1-rtTA transgenic mice: a tool for inducible gene expression in hair cells of the inner ear. Sci. Rep. 2014; 4, 6885. CrossRef
46. White PM, Doetzlhofer A, Lee YS et al. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature 2006; 441, 984–7. CrossRef
47. Chen Y, Yu H, Zhang Y et al. Cotransfection of Pax2 and Math1 promote in situ cochlear hair cell regeneration after neomycin insult. Sci. Rep. 2013; 3, 2996. CrossRef
48. Yu Q, Wang Y, Chang Q et al. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014; 21, 71–80. CrossRef
49. Takada Y, Beyer LA, Swiderski DL et al. Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear. Res. 2014; 309, 124–35. CrossRef
50. Askew C, Rochat C, Pan B et al. Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 2015; 7, 295ra108. CrossRef
51. Husseman J, Raphael Y. Gene therapy in the inner ear using adenovirus vectors. Adv. Otorhinolaryngol. 2009; 66, 37–51. CrossRef
52. Adunka O, Unkelbach MH, Mack M et al. Cochlear implantation via the round window membrane minimizes trauma to cochlear structures: a histologically controlled insertion study. Acta. Otolaryngol. 2004; 124, 807–12. CrossRef
53. Skarzynski H, Lorens A, Piotrowska A et al. Preservation of low frequency hearing in partial deafness cochlear implantation (PDCI) using the round window surgical approach. Acta. Otolaryngol. 2007; 127, 41–8. CrossRef
54. Irving S, Gillespie LN, Richardson RT et al. Electroacoustic stimulation: Now and into the future. Biomed. Res. Int. 2014; 350504. CrossRef
Affiliations
Patrick J Atkinson
Stanford University, School of Medicine, Department of Otolaryngology,
Stanford, CA, USA
Madeline Nicholson
Bionics Institute, 384 Albert Street,
East Melbourne, Victoria, 3002, Australia
Rachael T Richardson
Bionics Institute, 384 Albert Street,
East Melbourne, Victoria, 3002, Australia and
University of Melbourne,
Department of Otolaryngology,
Melbourne, Australia