Drivers and methodologies for making cell therapy process changes
Cell Gene Therapy Insights 2016; 2(1), 85-94.
10.18609.cgti.2016.003
Cell therapies are a new and exciting modality with the potential to treat a variety of severe unmet medical needs. Although the types of cell therapies currently in clinical development encompass a wide range of cell types with various functions, they all can be classified as highly complex biological products that are challenging to characterize. As is the case with all complex biological products, the processes by which they are produced, to some degree, define the product such that process changes must be highly scrutinized before they can be implemented. In order to make process changes for a cell therapy product, one must employ a combination of strong analytical tools, a thorough understanding of how the cell is intended to function as a therapy, detailed process characterization which elucidates how process parameters affect product attributes, and sound engineering principles to understand how the chemical and physical parameters of the process can be controlled to yield a comparable product after the process change.
Submitted: January 7 2016 Published: March 21 2016;
Throughout the clinical development of any compound, process changes are inevitable. In today’s competitive environment and with patients’ health and lives at stake, it is not practical to invest the time and resources to design a commercially desirable process for initial clinical studies. It makes much more sense to deliver a product to the clinic quickly to determine its safety and proof of activity so that a decision can be made as to whether to further develop it. This approach allows more treatments to be tested in the clinic more quickly but will often result in the need for process changes as a product moves closer to commercialization. In many cases, the optimum balance of speed and risk requires accepting that some changes will be required throughout clinical development. Many of these can be identified and planned in advance.
Drivers for process changes
The key reasons for making process changes are to ensure or improve product quality in terms of safety and efficacy, to improve process capacity to meet clinical and commercial demands, and to reduce costs. Product safety and efficacy are the most critical considerations from the time of product concept and initial process design, all the way throughout the product’s life cycle. An improved product understanding through in vitro studies, animal studies, translational efforts, and most relevantly, through clinical data, as the product is developed, typically yields information as to how to improve product safety and efficacy. Process characterization efforts, which provide information on how process input parameters affect product quality attributes, are also critical. These efforts provide information on how to improve process robustness and reproducibility, which in turn yields a more consistent product.
Efforts to increase process capacity are nearly always required as clinical trials progress towards licensure and commercial demands typically far exceed what is required for clinical development. By nature, clinical trials will include more patients as trials move from Phase I to Phase III and beyond. Product dose often increases as well, with cell therapy development strategies often employing a dose escalation protocol in an early trial to find the maximum tolerated dose. Once it is reached, it is very likely that the remainder of that trial, as well as subsequent trials, will utilize the highest practical dose. Commercial needs are forecasted early in development to inform process development as to what scale the final process should be. These marketing forecasts are updated as more information becomes available throughout clinical development. Such information could include more defined patient populations who will be treated, based on clinical data from one’s trial, as well as a better understanding of the product’s projected market share, based on an improved prediction of the competitive landscape, which is usually rapidly evolving. Ultimately, it is of critical importance to have a process and a manufacturing methodology that can meet commercial demand. It would be a major disservice to the patients we serve if we have a product which can improve their quality of life, but which we cannot provide to all patients because we cannot produce enough of it.
Product manufacturing cost is typically not a major consideration in early product development because, as discussed earlier, the best strategy is often to test a product’s proof-of-concept quickly to determine if it is worth developing, before investing a great deal of resources in it. Of course, early product costs cannot be so high that the initial pre-clinical studies and Phase I trials are not affordable. Once a product moves past Phase I, its cost of goods (COGs) typically become a high priority. The reason for this is that one needs to be able to commercially make the product at a low enough cost to yield a profit that offsets the enormous development expense. As is the case in any other industry, if a product is not profitable it doesn’t make business sense to develop it. The development expenses of a cell therapy product or of any pharmaceutical product, with a strong probability of failure during development, must also be considered in the context of all the products that never make it to commercialization and whose expenses cannot be recouped.
Product COGs are most greatly influenced by labor, testing, and raw material costs. Labor can be reduced via efforts to change process unit operations to ones that rely more on automation and less on human input. Moreover, unit operations that can be scaled up to yield more product per step or per batch will yield lower COGs since the costs of manufacturing will be spread across more product. Streamlining process steps to reduce manpower needs is another way to reduce COGs. Testing costs will be reduced similarly to labor costs as processes are scaled up and more product is made per batch, allowing testing costs to be spread across more product. Raw material costs will typically decrease via economies of scale as more product is made; however, the costs of many raw materials used to make cell therapies, such as animal- and human-sourced raw materials and recombinant proteins will still be inherently very high. Less expensive alternatives can be sought but, as is the case with process changes that reduce labor or optimize the process in some other way, these changes will need to be shown to yield a comparable product.
Process changes are ideally made proactively to meet clinical needs and to maintain a commercial line-of-sight, but often times process changes are reactive as problems are encountered or as opportunities arise. Product knowledge and process understanding are always increasing as process development data are being amassed and as clinical product is being produced. As more clinical product is generated and more process development experiments are performed, it is quite common for new challenges to arise, which must be addressed through process changes or modifications. Similarly, as the field progresses, new opportunities present themselves that could warrant a process change. For example, a few years ago recombinant enzymes with a similar function as trypsin became commercially available. Before these were available, porcine trypsin was universally used to remove cells from surface substrates upon which the cells were grown. While the porcine trypsin could be sourced and tested to satisfy regulatory and safety requirements, recombinant versions that were produced without any animal components offered a simpler, lower-risk alternative. Therefore, a movement towards these alternatives commenced. Overall, as a product moves from pre-clinical development to commercialization, one must not only be forward looking to make process changes, which will help meet increasing clinical demands and yield a more favorable commercial product, but also be ready to address problems that require process changes and to take advantage of opportunities to make a better process and higher quality product.
Timing for Process Changes
Process changes are easier to justify, from a regulatory point of view, earlier in development but increasing process and product knowledge and improved analytical tools, which enable such changes, are available later in development (Figure 1). When a product is early in clinical development for example, before Phase II clinical trials, it is typical that only its safety has been demonstrated in humans. Therefore, the regulatory burden on the clinical trial sponsor, when making process changes, is to prove that the processes utilized before and after the change result in a comparable product, in regard to its safety profile. The risks of comparable efficacy and potency are, of course, business risks that the clinical sponsor must weigh against the benefits of the process change. These would also be regulatory risks if clinical data showing efficacy already exist. As clinical data become available as trials progress and as efficacy is demonstrated, the tolerability for process changes will inherently decrease. Hence, both major process changes and minor process improvements become less and less common as the product moves further through clinical development.
This decreased tolerability for change, as the product moves through clinical development, doesn’t mean changes cannot be made. In fact, changes will have much greater data-driven support later in development. This is due to a number of factors. Product knowledge is ever increasing as clinical trial information, translational and in vivo animal data, and mechanism of action understanding evolves. Process understanding is also increasing as process characterization experiments are carried out. These types of studies demonstrate the effect of process input parameters on key product attributes. As these data are amassed, they allow process development to accurately predict what ranges of process input parameters are acceptable, thereby providing process flexibility and, in some cases, enabling process changes. A simple example of process characterization data is depicted in Figures 2a and 2b. Figure 2a shows what the typical growth curve might look like for a culture-expanded cellular therapy product. Having more cells at the end of the process is generally desirable due to economics but it might also be desirable for the cells to be in exponential phase at the time of harvest. Therefore, Figure 2a depicts a possible desired range for harvest density. Figure 2b shows the effect of seeding density on harvest density for four specific seeding densities examined under controlled conditions. The selected range, shown in Figure 2b, might be chosen to provide flexibility when seeding the culture expansion step. There is a minimum seeding density that yields a high enough harvest density for favorable economics but a maximum seeding density beyond which there is really no benefit but which would waste cells during seeding.
Further process data are also generated through clinical trial supply efforts. Product is manufactured for clinical trials typically using the best process available at the time, with process input parameters targeted for their optimal values. Trending and tracking of product data when a process is run the same way many times provides important information about process variability. Although clinical production efforts are often successful in running the process the same way repeatedly, process parameters, in reality, often experience deviations. The effects of these deviations also provide valuable data, much the same way that process characterization experiments provide data as to how process inputs affect process outputs.
At the same time that process understanding is increasing, analytical tools are being developed to better understand the product in a number of ways. First, assays that were available at the start of development are being improved. An increasing understanding of the effects of the assays inputs on the assays outputs can be used to improve assay reproducibility and robustness. Furthermore, making changes to the assay methods can enable measurement of more relevant product attributes. Another way to achieve this latter goal is through new assays, which are often being developed throughout clinical development. For example, as product mechanism of action is elucidated through translational efforts and in vivo animal studies, potency assays, which represent product efficacy, can be developed.
This increasing base of product knowledge (how the product behaves in terms of safety and efficacy), process characterization (how process input parameters affect key product attributes), and analytical tools (knowing what is relevant to measure and having more confidence that you are measuring it accurately) together allow one to make process changes even when the regulatory burden can be high.
So when should and shouldn’t process changes be made? Both major process changes and minor process improvements can be relatively freely implemented before product is generated for key investigational new drug (IND)- enabling animal studies. Since product data generated to that point will either not be used to justify its safety and activity or is only supplementary to the data set to be used to do so, the risk that process changes will affect the product is only a business risk. Moreover, the risks are probably very low at that point since product knowledge is likely to be very low. Once a process has been defined for the key IND-enabling animal studies, and in particular for the IND-enabling safety studies, no major process changes should be introduced for making product for the Phase I clinical trial. The reason for this is that the product is being administered into humans for the first time and its expected safety is predicated on the animal data in the IND. Any changes to the process that might even remotely have a chance of affecting product safety will not be tolerated. Once a Phase I trial has been completed and safety has been established in humans, changes are much more tolerable, although a clear rationale must be presented as to why changes will not affect product safety. Therefore, the ideal time to make major process changes is often between Phase I and Phase II studies or between different Phase II studies, when risks of changes are still not very high, when product and process knowledge are increasing, and when better analytical tools are available. Although it makes sense to implement changes between clinical trials, major process changes should be avoided during a clinical trial so as not to confound the clinical results with the variable of what might be a different product being administered to different patients.
Once clinical efficacy is established and there is justification for a Phase III trial, process changes will be very difficult to make. The risks of making a change that will affect product efficacy now becomes a regulatory risk as well as a business risk since a clinical sponsor should only be allowed to administer a product to such a large number of patients as will be in a Phase III trial when there is a strong reason to believe there will be a clinical benefit. Therefore, the data and rationale to support a change between Phase II and Phase III, and of course after Phase III or product licensure, must be extremely strong.
Although process changes are typically not implemented at these later stages of clinical development, process scale up, equipment changes, and facility changes can be acceptable. If process scale up is absolutely required to enable the production of sufficient supply to meet patient needs, as it often is when progressing from Phase II to Phase III and beyond Phase III, it can be implemented. Applying solid engineering principles usually enables one to scale up a process that yields comparable product but the data still need to be generated to support the scale up. Equipment and facility changes would be more difficult to justify but if the drivers for change and the data supporting the change are strong, then these are possible even at these later stages of clinical development.
Methodology for Making Process Changes
Once the drivers for a particular process change are clear, data must be generated to enable a clear understanding of the effects of this change on the product. Most importantly, it must be demonstrated that the product still meets its release specifications after the process change. What else is needed really depends on the change. There are typically a number of other assays to choose from, beyond what have been selected as release assays. Table 1 includes the types of cellular characteristics that may be important for the product’s intended function as well as the general types of assays one might use to measure these. The decision as to which assays to use to assess the effects of the change requires the application of one’s product knowledge to understand how the product is intended to function in vivo and how the process change might affect those functions. It is not expected to utilize all of these to assess every change and certainly one would only be expected to perform in vivo or ex vivo studies to justify changes that cannot be assessed with in vitro measures alone.
| Table 1. Cellular characteristics and general assay methods relevant to cell therapies. | |
|---|---|
| Cellular characteristics | General assay methods |
| Physical characteristics: cell number, size, morphology, viability | Cell-based assays |
| Biochemical properties: growth, metabolism, protein/gene expression & regulation | Flow cytometry |
| Cellular responses: potency, differentiation, migration, apoptosis/necrosis, signaling, secretion, cytotoxicity, antigen presentation | Biochemical, enzymatic & ligand binding assays |
| Molecular biology tools | |
| Omics: genome, proteome, miRNA | |
| Ex vivo | |
| In vivo animal, human |
Applying strong engineering principles is another important dimension to consider when implementing a process change. Understanding the effects of the chemical and physical parameters on the product when it is made by the original process as compared to the revised process can be critical in deciding how to evaluate the change. An example of how such engineering principles could be applied is the case of switching from a batch centrifugation step to a continuous centrifuge step for the washing of a cellular product after a cell culture step. In batch centrifugation, the product is transferred to closed centrifuge bottles or bags and placed in a standard centrifuge, whereas in continuous centrifuge, the product is flowed through a device with a counteracting centrifugal force. Although such an example may seem like a difficult process change to justify, a strong understanding of the effects of the physical parameters that the product experiences during these steps could make this change rather straightforward. First, one could characterize the range of centrifugal force that the product experiences in the batch centrifugation step and the time at which the product experiences that force. Next, one could show acceptable ranges for these parameters, either based on the effects of these on product attributes or by data mining the ranges used to previously produce acceptable clinical supply. Finally, one could then design the continuous centrifugation step so that the product experiences centrifugal forces in the same range that had been deemed acceptable in the batch centrifugation step. As for the fluid flow that the product encounters in the continuous centrifugation step but not the batch centrifugation step, one could utilize data from other steps of the post-culture process when product streams do undergo fluid flow. These data could be used to define acceptable ranges of, for example, shear stress on the product, and then the flow in the continuous centrifugation step could be controlled to yield shear stresses in this range. If there are other physical or chemical effects encountered in the continuous centrifugation step but not the batch centrifugation step, one could take a similar approach. Finally, the proper analytical methods would need to be applied to ensure that a comparable product is being produced when using continuous centrifugation as compared to batch centrifugation.
An example of how to apply both strong engineering principles and relevant analytical methods when implementing a process change is Celgene Cellular Therapeutics’ switch of a final product container from a 20 mL cryopreservable bag to a 20 mL cryopreservable vial. This container is used for the long-term storage of our placental-derived, mesenchymal stromal-like, IV infusible cellular product, PDA-001. After cell growth and harvest, cells are washed of residual cell culture medium components and buffer exchanged into the final formulation. Final containers are filled and temperature is decreased using a controlled rate freezer before long term storage in the vapor phase of liquid nitrogen (-175°C). The controlled rate freezer chamber temperature had been controlled in a manner to yield a cryobag freezing profile which had best preserved cellular viability and function through the freezing and subsequent thawing of product, which occurs right before patient administration.
As a first pass, this same controlled rate freezing protocol was applied to freezing product in cryovials, as had been used when freezing product in cryobags. Figures 3a and 3b show the effects of the original freezing protocol on trypan blue viability and cellular potency for product in bags (x-axis) vs product in vials (y-axis). Figure 3a shows that trypan blue viability of product frozen in vials is always within 5% of that for product frozen in bags. Moreover, other analytical methods used for this product showed no differences for product frozen in vials as compared to product frozen in bags (data not shown). However, Figure 3b shows that potency of product frozen in 20 mL vials is, in most cases, more than 30% lower than potency of product frozen in bags. As a comparison, potency of product frozen in 2 mL vials is always within 10% of potency of product frozen in bags. This suggests that the potency differences between bags and vials were caused by the different geometrical configurations of the bags compared to vials rather than the different product-contacting materials.
A close examination of the freezing profiles of the 20 mL bags and the 20 mL vials (Figure 4) showed that due to the increased heat of fusion duration experienced in the vials, between approximately 40 to 70 minutes into the freezing procedure, the vials experienced more than a two-fold increase in maximum cooling rate between approximately 70 and 100 minutes. Separate experiments to characterize the effects of this cooling rate on potency showed that increasing freezing rates directly caused a decrease in product potency (data not shown). The controlled rate freezing protocol was subsequently re-designed with a different chamber temperature profile to account for this increased heat of fusion duration experienced in the vials and to yield an acceptable maximum cooling rate. The results of this revised freezing protocol are shown in Figures 5a and 5b. As was the case with the original freezing protocol, Figure 5a shows that trypan blue viability of product frozen in vials is always within 5% of that for product frozen in bags. In fact, the difference between viability for product in vials as compared to bags is even less than it was with the original freezing protocol. Furthermore, Figure 5b shows that the difference in potency for product frozen in vials is generally within 10% of the potency of product frozen in bags.
This example shows how relevant analytical methods can allow one to detect an undesirable effect of a process change and how solid engineering methods and a good understanding of the physical effects of the process on the product can enable one to design a process change so that a comparable product is obtained. If the relevant analytical methods were not available, it might have been concluded that the process could be run with the original freezing protocol and the undesirable effects on the product wouldn’t have been detected.
Conclusions
When making process changes for a complex biological product such as a cellular therapy, the proper resources must be invested to demonstrate that the product made after the process change is comparable to the product made before the process change. A combination of relevant analytical tools, detailed process characterization to understand how process inputs affect product attributes, a thorough understanding of the function of the cells and how chemical and physical parameters affect the cells, along with a sound engineering approach to design the right process, together enable smart process changes. When deciding whether a process change can be implemented, the benefit of the change, in terms of product quality, process capacity and product costs must always be weighed against the risks on product safety and efficacy. The timing of the change, in terms of the stage of clinical development, will often factor into this decision and how much risk can be accepted. In the end, however, the burden is always on the clinical sponsor to demonstrate via data and strong science that the process change yields a product that is comparable in terms of its safety, efficacy, and quality.
Figures
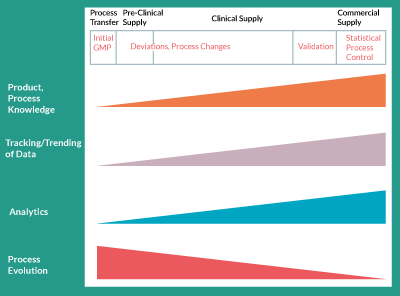
Figure 1. Increasing knowledge, data, and analytical tools but decreasing tolerability for changes through development.
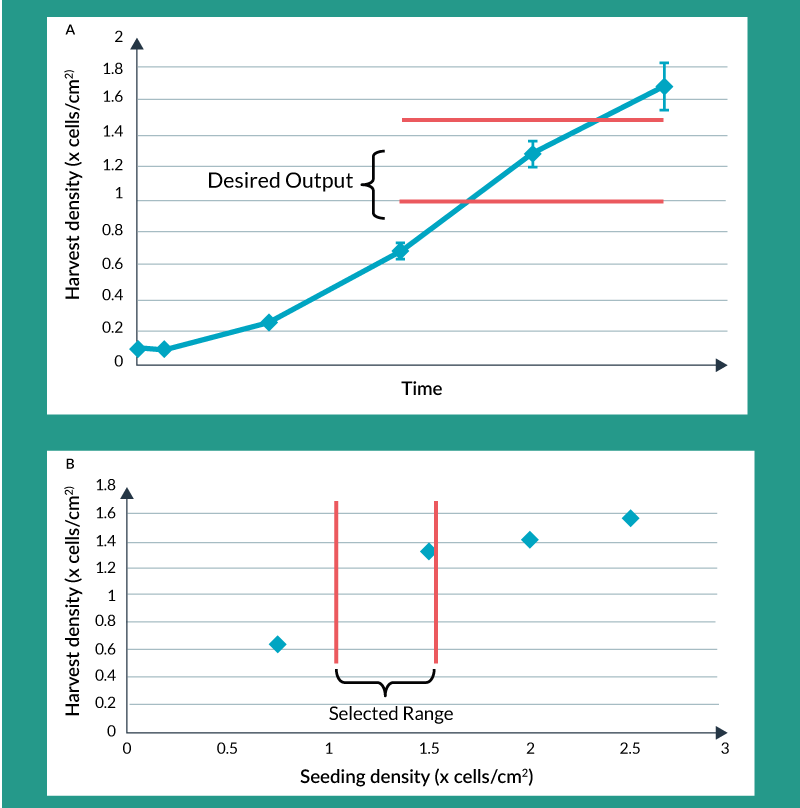
Figure 2. Process characterization: optimizing seeding density for maximum yield. A) Growth Curve B) Harvest Density vs Seeding Density
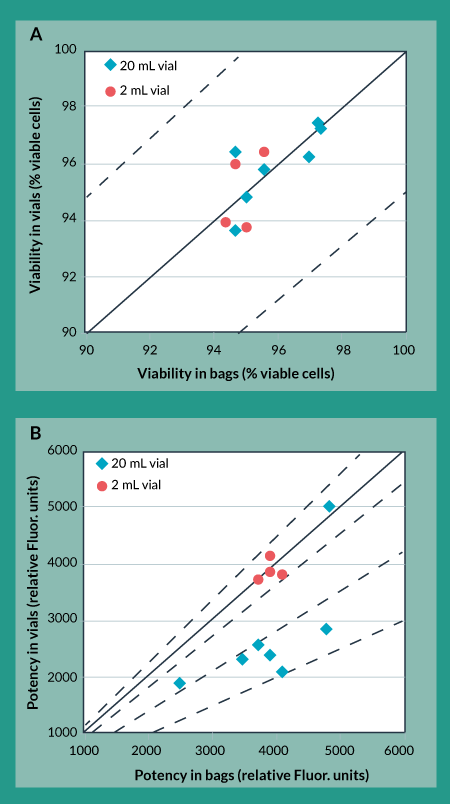
Figure 3. Cryovial to cryobag comparison using original freezing protocol. A) Trypan Blue Viability B) Potency.
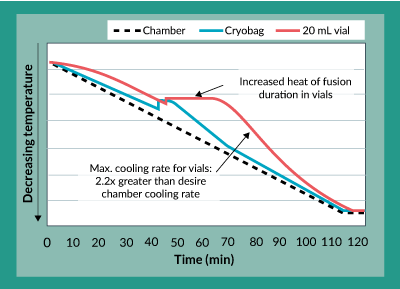
Figure 4. Temperature profile comparison for cryovials and cryobags using original freezing protocol.
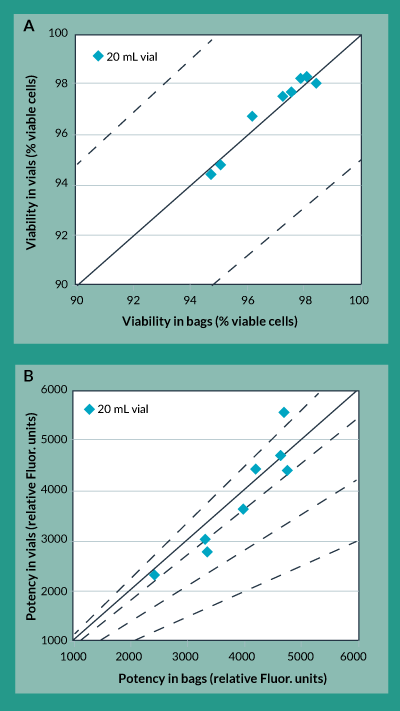
Figure 5. Cryovial to cryobag comparison using revised freezing protocol. A) Trypan Blue Viability B) Potency
Financial & competing interests disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Affiliations
Gregory Russotti, PhD
Vice President, Technical Operations
Celgene Cellular Therapeutics,
7 Powder Horn Dr, Warren,
NJ 07059, USA
grussotti@celgene.com