Beta testing: preclinical genome editing in β-globin disorders
Cell Gene Therapy Insights 2015; 1(2), 231-242
10.18609/cgti.2015.021
The β-globin disorders β-thalassemia and sickle cell disease have been at the forefront of gene therapy development from its very inception. Owing to their frequency, severity and exceptionally well characterized molecular pathology, and to the availability of hematopoietic stem and progenitor cells as substrate for therapies, these disorders promise both fast insights into new methodologies and eventual return on investment. Accordingly, β-globinopathies are also a favorite subject of the nascent field of genome editing and its most pioneering approaches to achieving therapeutic functional correction of gene expression. Be it by mutation-specific correction, modulation of disease modifiers or site-specific gene addition, genome editing of β-globinopathies has already delivered significant insights into the design of synthetic nucleases and the suitability of different correction strategies. This Expert Insight reviews recent progress in the application of gene-editing tools and different model systems towards the establishment of new therapies for β-globin disorders.
Submitted for review: Oct 5 2015 Published: Dec 10 2015
β-globinopathies: A continuous test bed for gene therapy approaches
The β-globinopathies as recessive monogenic disorders of the hematopoietic system were infamously the first targets for gene therapy more than 35 years ago [1], and recently β-globin was once again a controversial “first” in the application of gene editing to human zygotes [2]. Contributing factors to the ongoing effort by a plethora of researchers and companies to use β-globinopathies as a benchmark for new gene therapy approaches are: high disease prevalence, high natural childhood lethality, easy accessibility of autologous hematopoietic stem and progenitor cells (HSPCs) as gene therapy substrate, established efficacy of allogeneic HSPC transplantation and the small size and exemplary characterization of the regulation of HBB gene expression. Hope for a universal cure for β-globinopathies now focuses on gene editing approaches, not only because of recent technical developments, but also for reasons of safety and efficacy. After all, permanent gene addition using integrating vectors has the inherent risk of insertional mutagenesis [3], and for severe forms of β-globinopathies struggles to be fully therapeutic, owing to size restrictions on the transgene and the high transgene expression levels required [4].
The main adult hemoglobin (HbA) is a heterotetrameric metalloprotein, containing two α- and two β-globin chains. In sickle cell disease (SCD), a HBBE6V missense mutation results in toxic precipitates of HbS tetramers, containing the sickling β-globin (βS), while in β-thalassemia absent or diminished expression of β-globin results in toxic homotetramerization of surplus α-globin. The pathophysiologies of both β-thalassemia [5] and SCD [6] have been reviewed in detail elsewhere; suffice it to say that the above intracellular toxicity leads to vaso-occlusion and painful crises for SCD and to (myeloid) ineffective erythropoiesis and (peripheral) hemolysis for both disorders, with wide-ranging systemic and multi-system effects.
An important difference between β-thalassemia and SCD when approaching the development of a new therapy is a catalogue of over 340 primary mutations for the former and a single mutation for the latter [7,8]. This limits commercial interest in developing mutation-specific cures for β-thalassemia, also because many patients are compound heterozygotes for different β-globin mutations, including β-thalassemia/SCD.
As a key modifier of disease severity, α-globin has been considered as a therapeutic target for amelioration of β-thalassemia [9]. However, long-standing genetic and recent experimental evidence highlight the β-like fetal γ-globin in particular as a key ameliorating factor that might prove fully and universally therapeutic for both β-globinopathies [10]. γ-globin replaces β-globin as a binding partner for α-globin in a fetal hemoglobin (HbF) heterotetramer, and in SCD prevents HbS precipitation and sickling of erythrocytes. Expression of γ-globin is controlled by a still-expanding network of loci, proteins and micro RNAs [4], with γ-globin repressors KLF1 and BCL11A [10] and their response elements as promising therapeutic targets, in addition to the β-globin locus itself.
As gene editing tools, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats linked to Cas9 (CRISPR/Cas9) RNA-guided nucleases (RGNs) have to date been applied to β-globinopathies (see review by Bassett in this spotlight issue [11]), as well as triplex-forming peptide nucleic acids. Figure 1 illustrates gene editing approaches currently employed to β-globinopathies, with Table 1 summarizing the corresponding targets and efficiency data as detailed below.
| Table 1. Genome-editing achievements in β-globinopathies. Reports are listed first by target sequence (SCD mutation, β-thalassemia mutations in their order in HBB; HPFH mutations in their order in HBG1/2, BCL11A target regions), then by their order of appearance in the main text. All iPSCs are of human origin. | ||||
|---|---|---|---|---|
| Target [HGVS name] | Nuclease | Model | Reported Efficiencies | Ref. |
| βS [HBB:c.20A>T] | ZFN | K562 | NHEJ 2%, HDR 0.2% bulk HDR 95% clonal | [14] |
| TALEN | K562 | HDR 13% bulk (19% of transfected cells) | [15] | |
| TALEN | iPSC | HDR 63% clonal | [16,17] | |
| ZFN | iPSC | HDR 67% (2 out of 3) clonal | [18] | |
| ZFN (x3) | HEK293T | NHEJ 12.9%, 2.5% & 1.2% (HEK293T) bulk | [19] | |
| iPSC | HDR 33.3%, 37.9%, 26.3% (Hs iPSC) clonal | |||
| RGN, TALEN | iPSC | HDR 33% (TALEN), 12.3% (RGN) clonal | [22] | |
| RGN, TALEN, ZFN | HEK293T | NHEJ 45% (HEK293T), 3.2–3.6% (Hs iPSC) bulk, RGN | [23] | |
| ZFN | Hs mPB & BM CD34+ | HDR 18.4±6.7% (BM CD34+) bulk | [24] | |
| β -28 (A>G) [HBB:c.-78A>G] | RGN | iPSC | HDR 33% clonal | [21] |
| β CD 14/15 (+G) [HBB:c.45_46insG] | RGN | HEK293T | NHEJ 52% (zygote) | [13] |
| Hs zygotes | HDR 48.3% (HEK293T) & 7.4 % (zygote) bulk | |||
| β CD 17 (AAG>TAG) [HBB:c.52A>T] | RGN | HEK293T | NHEJ >50% (HEK293T) | [13] |
| Hs zygotes | ||||
| β CD 41/42 (-TTCT) [HBB:c.124_127delTTCT] | RGN | HEK293T | NHEJ undetectable (HEK293T) | [13] |
| Hs zygotes | ||||
| TALEN | iPSC | HDR 40% clonal | [20] | |
| RGN | iPSC | HDR 42% clonal | [21] | |
| β IVS2–1 (G>A)[HBB:c.315+1G>A] | TF-PNA | TG CHO | HDR 0.4% (CHO), PCR+ (K562 & CD34+) bulk | [54] |
| K562 | ||||
| Hs mPB CD34+ | ||||
| β IVS2–654 (C>T) [HBB:c.316–197C>T] | TALEN | iPSC | HDR 68% clonal | [20] |
| RGN, TALEN | iPSC | HDR 33% (TALEN) & 12.3% (RGN) clonal | [22] | |
| Aγ -175 T>C HPFH [HBG1:c.-228T>C] | TALEN | K562 | Aγ promoter activity x2 (K562 & MEL) & β-globin promoter activity x0.5 (MEL) clonal | [33] |
| TG MEL | ||||
| Aγ -117 G>A HPFH [HBG1:c.-170G>A] | TF-PNA | TG Mm BM | HDR 1.63% (CD34+) bulk | [34] |
| Hs mPB CD34+ | ||||
| BCL11A h +55, +58, +62 [NC_000002.11:g.60718000–25500] | RGN (x533) | HUDEP-2 | 40% HbF (4x control level), | |
| Hs CD34+ | 40% F cells (2x control level) bulk (CD34+, for sgRNA-1621) | [31,40] | ||
| BM: bone marrow; bulk: population-wide percentages after treatment, without enrichment or antibiotic selection of corrected cells; CHO: Chinese hamster kidney cell line; clonal: percentages amongst pre-selected antibiotic-resistant clones; HDR: homology-directed repair; HUDEP-2: human CD34+ HSPC-derived erythroid precursor cell line; HEK293T: SV40 T-antigen+ human embryonic kidney 293 cell line; iPSC: induced pluripotent stem cell; Hs: Homo sapiens; MEL: mouse erythroleukemia cell line; mPB: mobilized peripheral blood; NHEJ: non-homologous end joining; PCR+: too low to quantify, but detectable by PCR; sgRNA: synthetic guide RNA; TG: transgenic; zygotes: tripronuclear zygotes. | ||||
Horses for courses: correcting the primary mutation
Intuitively, correction of the primary mutation is the approach of choice for β-globinopathies, as it restores the exact genotype of normal or at least carrier individuals (Figure 1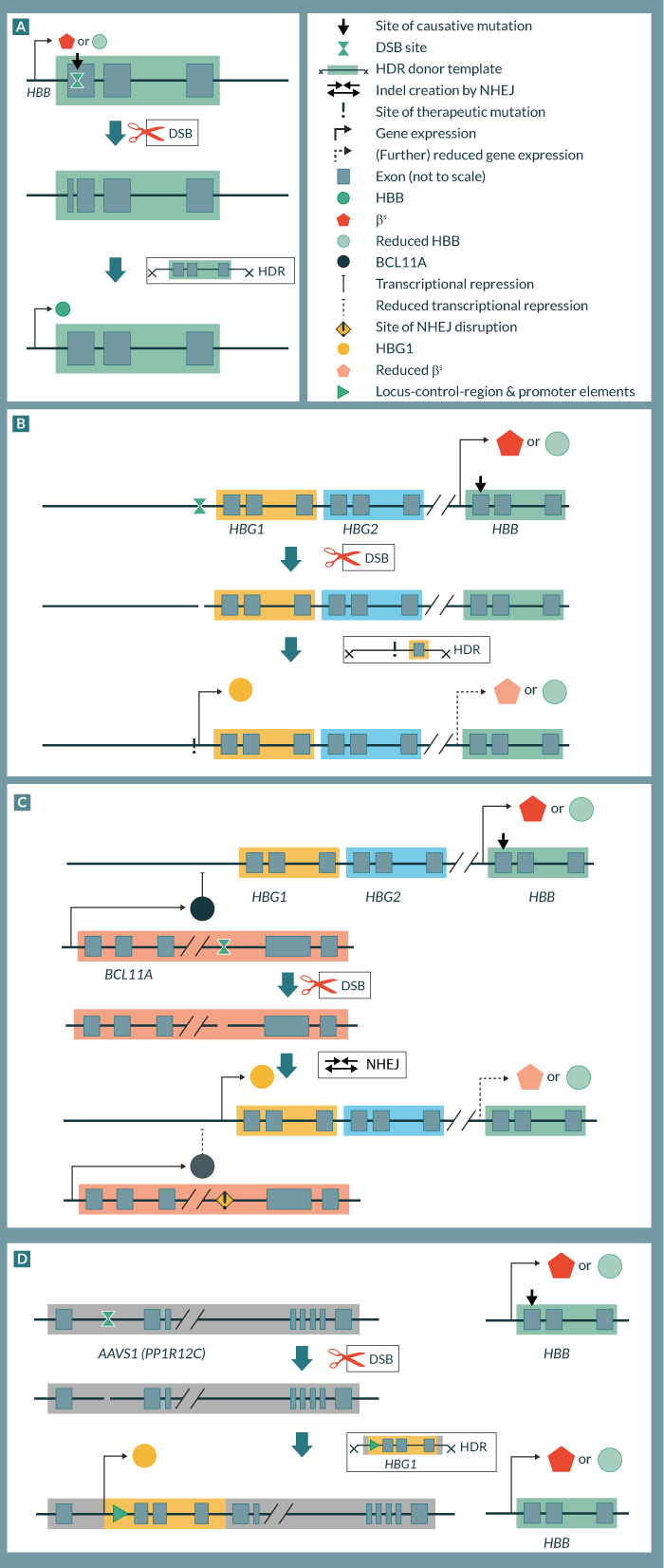
To date, all mutation-specific approaches to gene-editing of β-globinopathies have employed HDR-based sequence repair. As the model system furthest from clinical application, human erythroid K562 cells were edited with ZFNs and TALENs to target the βS site, giving 0.2% [14] and 13% [15] population-wide HDR efficiency, respectively. Low efficiency for the best out of four ZFNs tested in the former study may serve to illustrate the difficulty of identifying designer nucleases with therapeutic potential based on the evidently powerful ZFN technology. In preference to cell lines, from 2011 many studies started employing early-generation induced pluripotent stem cells (iPSCs) and ZFNs to target the βS mutation [16–19]. They did so with varying and low population-wide efficiencies, necessitating clonal selection of antibiotic-resistant clones to quantify and analyze intended recombination events and raising doubts over the applicability of the technology to primary HSPCs [14].
Mindful of a likely need for enrichment towards clinical application of genome editing, subsequent efforts for the β-globinopathies emphasized the fidelity and full erythropoietic potential of iPSCs, employing episomal [20] or footprintless piggyBac vectors [17,21,22] for reprogramming. One such study applied TALENs to SCD and demonstrated the absence of off-target cleavage for predicted sites after correction [17]. Similarly, Ma et al targeted two β-thalassemia mutations using TALENs and employing a universal full-length-HBB donor sequence for correction [20]. Establishing that efficient designer nucleases are repurposable for different β-thalassemia mutations, Xie et al used a single RGN to cleave HBB intron 1 and achieved repair of two independent mutations, one in the promoter and one in exon 2 of HBB in iPSCs of a compound heterozygous β-thalassemia patient [21]. For cells corrected for either mutation the study then confirmed erythropoietic differentiation and restoration of HBB mRNA expression. Using RGNs in comparison to previously published ZFNs and TALENs, Huang et al achieved remarkable population-wide correction efficiencies in patient-derived iPSCs (3.2–3.6%), followed by efficient in vivo erythroid differentiation and enucleation of corrected cells [23]. Moving even closer towards clinical application of SCD gene editing, Hoban et al applied ZFNs to mobilized peripheral-blood (mPB)- and BM-derived CD34+ cells and achieved over 18% population-wide correction efficiency [24]. Despite 45% donor chimerism in transplanted NOD-scid IL2Rgnull (NSG) mice 5 weeks after treatment, only low levels of long-term engraftment were achieved, possibly indicating a recalcitrance of long-term repopulating cells to editing conditions [25].
Several studies of the thalassemias focused on the HBBIVS2–654 splice-site mutation common in Asian populations. Targeting by a monomeric TALE–endonuclease fusion showed lowered genotoxicity compared to its dimeric TALEN counterpart, albeit at reduced efficiency [26]. In an independent study, RGN-mediated correction improved hematopoietic differentiation in patient-derived iPSCs with no detectable cleavage at predicted off-target sites [27]. Comparing the efficiency of TALENs and RGNs for the same locus, Xu et al demonstrated higher efficiency and lower off-target activity of TALENs, erythroid differentiation and the presence of normal HBB mRNA [22]. Another study used HBBIVS2–654-specific ZFNs to compare the relative impact of iPSC reprogramming and ZFN-mediated gene correction on genome integrity. Using stringent quality filtering after exome sequencing, it determined that gene editing created four times the number of indels and five times the number of non-synonymous nucleotide variations, emphasizing the emerging consensus that each specific designer nuclease will need genome-wide off-target assessment as a component of its preclinical validation [28].
Jack of all traits: activating γ-globin
Therapies covering both β-thalassemia and SCD are the commercial ideal for therapeutic development, and for gene editing approaches this notion is currently synonymous with re-activation of endogenous γ-globin. Unsurprisingly, different strategies for γ-globin reactivation are therefore a focus not just of academic, but also of IP and commercial R&D activity, much of which might still be undisclosed [29–32]. With often low population-wide correction efficiencies and uncertainties over the γ-globin level achievable in each edited cell, it is still unclear whether currently employed re-activation strategies that are potentially suitable for all β-globinopathies will in the end be therapeutic for any of them.
An HDR-based strategy for reactivation of γ-globin was pursued by Wienert et al, who used TALENs to re-create the natural Aγ-175(T>C) hereditary persistence of fetal hemoglobin (HPFH) mutation (Figure 1B), albeit in erythroid cell lines and with low γ-globin induction efficiency [33]. An earlier attempt to introduce the Aγ-175(T>C) HPFH mutation in human mPB CD34+ cells utilized triplex-forming peptide nucleic acids and achieved a remarkable but clinically negligible 1.63% population-wide HDR efficiency [34]. Importantly, HSPCs mainly employ non-homologous end joining (NHEJ) for double-strand-break repair, in part because HDR is favored in actively cycling cells [25], underlining the advantage of NHEJ-based strategies for reactivation of γ-globin, particularly in HSPCs.
Of conceivable strategies using NHEJ, the reproduction of highly therapeutic HPFH deletion mutations by two independent double strand break events is possible, but would likely suffer from lowered efficiency and uncontrollable recombination events [35]. Complete deactivation of γ-globin suppressors BCL11A or KLF1 by a single NHEJ event would be efficient but detrimental to the hematopoietic and erythropoietic compartment, respectively [36,37]. However, disruption of their response elements in the γ- and β-globin promoters or of the erythroid enhancer element of BCL11A (Figure 1C) would predictably only affect the erythroid compartment and globin-gene expression [38]. As an added benefit of this approach, editing non-coding target sequences has a lower incidence of off-target cleavage in exonic regions, but care must be taken to avoid off-target hits in homologous regulatory sequences elsewhere in the genome [39]. As the corresponding regulatory elements are mostly defined through widely spaced genetic HPFH markers, Canver et al recently employed 533 tiling RGNs (over 700 including controls) to define key elements of the murine BCL11A enhancer [40]. In the process they identified one RGN of particular potency also in human HSPCs which achieved a population-wide level of 40% HbF and 40% γ-globin positive cells (four times and twice the level of controls, respectively), thus defining a new benchmark for likely therapeutic HbF induction and warranting further preclinical analysis.
Something old, something new: site-specific gene addition
The recent wedding of designer nucleases with entire genes or expression cassettes for site-specific integration also promises to be fruitful for the β-globinopathies (Figure 1D). Numerous existing gene-addition vectors and their corresponding efficiency and safety data can be borrowed for this approach of gene replacement at the disease locus or of gene addition at inert genomic loci. The latter strategy allows faster development of new therapies by following optimized blueprints for combining different gene-addition vectors with pre-existing designer nucleases. These would typically be highly optimized for target specificity and efficient HDR at inert genomic loci (safe-harbor sites), with the AAVS1 (PPP1R12C) locus as the most popular target for HSPC gene therapy [41]. Application of this approach to β-thalassemia was reported as early as 2011 [42], when it relied on iPSCs and fluorescence-based selection of clones carrying the β-globin transgene. In the wake of this study, site-directed insertion of an α-globin cassette for α-thalassemia used a similar methodology [43]. Since then, only an adenoviral-encoded TALEN, specific to the β-globin locus control region and displaying 50% targeting efficiency in a human cell line, is of direct interest to the β-globinopathies, as it may allow the introduction of donor sequences for HBB gene replacement [44]. More significantly, however, have been recent general technological advancements, such as the up to fivefold enhancement in human cells of HDR-based repair at the AAVS1 locus by adenovirus-mediated knockdown of NHEJ components [45]. Moreover, optimization of the safe-harbor approach for HSPCs reached 3–6% HDR in primitive to early progenitors and 0–9% in bone-marrow CD34+ cells in chimeric NSG mice long-term, thus approaching efficiencies of clinical interest [25]. While the specific study focused on X-linked severe combined immunodeficiency, the underlying methods and infrastructure are also available for β-globin gene addition by a lentiviral vector currently undergoing clinical trial [46,47].
Translation Insight
State of play
Genome editing potentially avoids the danger of insertional mutagenesis inherent to current gene-addition approaches, and faithful correction of primary mutations would moreover achieve normal β-globin expression levels in corrected cells. The technology is therefore likely to transform the clinical application of gene therapy for the β-globinopathies. All three major gene-editing platforms – ZFNs, TALENs and RGNs – have the potential to proceed to clinical trials, depending on safety and efficacy of each individual nuclease and the associated culture and treatment protocols. Presently, there remain impediments to clinical translation of the technology, namely off-target effects and low efficiencies of HDR events in HSPC bulk populations. The additional concern of iPSC safety is secondary to low efficiencies in HSPCs, which would be the preferred cell population for clinical application. Recent findings already indicate how safety and efficacy of gene editing might be improved towards a clinical application of the technology for β-globinopathies based on HSPCs.
Safety
Off-target sites are potentially widespread [13,28] and essentially unpredictable [48], representing a clear safety concern for clinical application. Gene editing applications will therefore predictably incorporate genome-wide sequence analyses in human model systems as a mandatory part of preclinical safety assessments for each gene editing tool. In parallel, advanced designs for improved specificity of designer nucleases [49–53] will be incorporated as standard methods at least for gene editing studies geared towards clinical application.
Efficiency
Cell-specific and population-wide correction efficiencies remain separate concerns for different types of application. While disruption of response elements reaches high population-wide efficiencies by reliance on NHEJ, data are as yet insufficient to judge if the resulting engraftment and γ-globin expression per corrected cell will indeed reach therapeutic levels. Conversely, HDR-dependent correction of primary mutations will be therapeutic for each corrected cell, but suffers from lower population-wide efficiency, as does HDR-based re-activation of γ-globin expression. Site-specific gene addition as a hybrid approach combines HDR with the constraints of transgene delivery typical of gene addition. Owing to safety and mutation-independence of optimized designer nucleases (where safe-harbor sites are targeted) and potential use of pre-existing vectors for gene addition, however, the approach will likely still have a future. Finally, for all HDR-based strategies recent findings indicate that modification of repair pathways and optimized tools and treatment protocols may eventually overcome low population-wide efficiencies, with already high levels of correction in human HSPCs and despite as yet low levels of long-term repopulation for corrected cells in NSG mice [24].
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
Acknowledgements
The authors thank Petros Patsali for critical review of the manuscript.
This work has received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 306201 (ThalaMoSS).

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1.Johnson RS. Gene transfer experiment in humans meets with scant approval. JAMA 1980; 244(19), 2139–40. CrossRef
2.Zhang Z, Chen Y, Sun X et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Biomed. Res. Int. 2015; 6(5), 363–72. CrossRef
3.Cesana D, Ranzani M, Volpin M et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014; 22(4), 774–85. CrossRef
4.Finotti A, Breda L, Lederer CW et al. Recent trends in the gene therapy of beta-thalassemia. J. Blood Med. 2015; 6, 69–85. CrossRef
5.Rund D & Rachmilewitz E. Beta-thalassemia. N. Engl. J. Med. 2005; 353(11), 1135–46. CrossRef
6.Steinberg MH. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. Scientific World J. 2008; 8, 1295–1324. CrossRef
7.Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M. IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLoS One 2014; 9(7), e103020. Giardine B, Borg J, Viennas E et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014; 42(Database issue): D1063–D1069. CrossRef
8.Mettananda S, Gibbons RJ, Higgs DR. alpha-Globin as a molecular target in the treatment of beta-thalassemia. Blood 2015; 125(24), 3694–701. CrossRef
9.Bauer DE & Orkin SH. Hemoglobin switching’s surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 2015; 33, 62–70. CrossRef
10.Bassett A. The promise of therapeutic
11.Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013; 41(20), 9584–92. CrossRef
12.Liang P, Xu Y, Zhang X et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015; 6(5), 363–72. CrossRef
13.Vannocci T, Kurata H, de la Fuente J, Roberts IA, Porter ACG. Nuclease-stimulated homologous recombination at the human β-globin gene. J. Gene Med. 2014; 16(1–2), 1–10. CrossRef
14.Voit RA, Hendel A, Pruett-Miller SM, Porteus MH. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014; 42(2), 1365–78. CrossRef
15.Sun N, Liang J, Abil Z, Zhao H. Optimized TAL effector nucleases (TALENs) for use in treatment of sickle cell disease. Mol. Biosyst. 2012; 8(4), 1255–63. CrossRef
16.Sun N & Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol. Bioeng. 2014; 111(5), 1048–53. CrossRef
17.Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011; 118(17), 4599–4608. CrossRef
18.Sebastiano V, Maeder ML, Angstman JF et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011; 29(11), 1717–26. CrossRef
19.Ma N, Liao B, Zhang H et al. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free beta-thalassemia induced pluripotent stem cells. J. Biol. Chem. 2013; 288(48), 34671–79. CrossRef
20.Xie F, Ye L, Chang JC et al. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014; 24(9), 1526–33. CrossRef
21.Xu P, Tong Y, Liu XZ et al. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in beta-thalassemia-derived iPSCs. Sci. Rep. 2015; 5, 12065. CrossRef
22.Huang X, Wang Y, Yan W et al. Production of Gene-Corrected Adult Beta Globin Protein in Human Erythrocytes Differentiated from Patient iPSCs After Genome Editing of the Sickle Point Mutation. Stem Cells 2015; 33(5), 1470–79. CrossRef
23.Hoban MD, Cost GJ, Mendel MC et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015; 125(17), 2597–604. CrossRef
24.Genovese P, Schiroli G, Escobar G et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014; 510(7504), 235–40. CrossRef
25.Lin J, Chen H, Luo L, Lai Y, Xie W, Kee K. Creating a monomeric endonuclease TALE-I-SceI with high specificity and low genotoxicity in human cells. Nucleic Acids Res. 2015; 43(2), 1112–22. CrossRef
26.Song B, Fan Y, He W et al. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015; 24(9), 1053–65. CrossRef
27.Ma N, Shan Y, Liao B et al. Factor-induced Reprogramming and Zinc Finger Nuclease-aided Gene Targeting Cause Different Genome Instability in beta-Thalassemia Induced Pluripotent Stem Cells (iPSCs). J. Biol. Chem. 2015; 290(19), 12079–89. CrossRef
28.Cost GJ, Gregory PD, Guschin D et al. 2015. Methods and compositions for treatment of a genetic condition (EP2890780A2) Sangamo Biosciences, Inc. [https://patents.google.com/patent/EP2890780A2].
29.Takeuchi R, Groudine MT, Stoddard BL, Bender MA. 2015. Compositions and methods for the treatment of hemoglobinopathies (US20150166969A1) Fred Hutchinson Cancer Research Center [https://patents.google.com/patent/US20150166969A1/en].
30.Orkin SH, Reik A, Urnov F. 2015. Nuclease-mediated regulation of gene expression (US20150132269A1) Children’s Medical Center Corporation; Sangamo Biosciences, Inc. [https://patents.google.com/patent/US20150132269A1/en].
31.Sangamo BioSciences I. 13 May 2015. Sangamo BioSciences Announces Joint Steering Committee Decision To Consolidate ZFP Therapeutic® Strategy For Hemoglobinopathies [http://investor.sangamo.com/releasedetail.cfm?ReleaseID=912987].
32.Wienert B, Funnell AP, Norton LJ et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat. Commun. 2015; 6, 7085. CrossRef
33.Chin JY, Reza F, Glazer PM. Triplex-forming peptide nucleic acids induce heritable elevations in gamma-globin expression in hematopoietic progenitor cells. Mol. Ther. 2013; 21(3), 580–87. CrossRef
34.Li J, Shou J, Guo Y et al. Efficient inversions and duplications of mammalian regulatory DNA elements and gene clusters by CRISPR/Cas9. J. Mol. Cell Biol. 2015; 7(4), 284–98. CrossRef
35.Yu Y, Wang J, Khaled W et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med. 2012; 209(13), 2467–83. CrossRef
36.Vinjamur DS, Wade KJ, Mohamad SF, Haar JL, Sawyer ST, Lloyd JA. Kruppel-like transcription factors KLF1 and KLF2 have unique and coordinate roles in regulating embryonic erythroid precursor maturation. Haematologica 2014; 99(10), 1565–73. CrossRef
37.Bauer DE, Kamran SC, Lessard S et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013; 342(6155), 253–7. CrossRef
38.Luo Y, Zhu D, Zhang Z, Chen Y & Sun X. Integrative Analysis of CRISPR/Cas9 Target Sites in the Human HBB Gene. Biomed. Res. Int. 2015; 2015, 514709. CrossRef
39.Canver MC, Smith EC, Sher F et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015; 527(7577),192–7. CrossRef
40.Lombardo A, Cesana D, Genovese P et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods 2011; 8(10), 861–9. CrossRef
41.Papapetrou EP, Lee G, Malani N et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011; 29(1), 73–8. CrossRef
42.Chang CJ & Bouhassira EE. Zinc-finger nuclease-mediated correction of alpha-thalassemia in iPS cells. Blood 2012; 120(19), 3906–14. CrossRef
43.Saydaminova K, Ye X, Wang H et al. Efficient genome editing in hematopoietic stem cells with helper-dependent Ad5/35 vectors expressing site-specific endonucleases under microRNA regulation. Mol. Ther. Methods Clin. Dev. 2015; 1, 14057. CrossRef
44.Chu VT, Weber T, Wefers B et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015; 33(5), 543–8. CrossRef
45.Roselli EA, Mezzadra R, Frittoli MC et al. Correction of beta-thalassemia major by gene transfer in haematopoietic progenitors of pediatric patients. EMBO Mol. Med. 2010; 2(8), 315–28. CrossRef
46.Miccio A, Cesari R, Lotti F et al. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc. Natl Acad. Sci. USA 2008; 105(30), 10547–52. CrossRef
47.Gabriel R, von Kalle C, Schmidt M. Mapping the precision of genome editing. Nat. Biotechnol. 2015; 33(2), 150–2. CrossRef
48.Miller JC, Zhang L, Xia DF et al. Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat. Methods 2015; 12(5), 465–71. CrossRef
49.Kleinstiver BP, Prew MS, Tsai SQ et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015; 523(7561), 481–485. CrossRef
50.Wyvekens N, Topkar VV, Khayter C, Joung JK, Tsai SQ. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum. Gene Ther. 2015; 26(7), 425–31. CrossRef
51.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014; 32(3), 279–84. CrossRef
52.Zetsche B, Gootenberg JS, Abudayyeh OO et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015; 163(3), 759–71. CrossRef
53.Chin JY, Kuan JY, Lonkar PS et al. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc. Natl Acad. Sci. 2008; 105(36), 13514–19. CrossRef
Affiliations
Dr CW Lederer* & Dr Marina Kleanthous
Cyprus School of Molecular Medicine & Department of Molecular Genetics of Thalassaemia, The Cyprus Institute of Neurology and Genetics, 6 International Airport Avenue, 1683 Nicosia, Cyprus.
*Author for Correspondence
Tel.: +357 22 392764
Fax: +357 22 392615
Lederer@cing.ac.cy