Stem cell-derived organoid cultures and genome editing tools
Cell Gene Therapy Insights 2015; 1(2), 243-251.
10.18609/cgti.2015.019
The term stem cell was first used in the late 19th century to describe ‘the ancestor’ unicellular organism, the origin from which all multicellular organisms evolved. Our definition today is that of a cell characterized by two properties: self-renewal (the capacity to generate new stem cells) and multipotency (the ability to differentiate into different cell lineages) [1–4]. Due to these characteristics, stem cells hold great potential for clinical use by providing an unlimited source of cells for cell therapy in regenerative and/or personalized medicine. To realize this potential, the development of stem cell culture systems, as well as stem cell genome editing tools, has been of paramount importance. Here, recent advances in culture systems and genome editing tools will be discussed. This Expert Insight will provide an overview of current, state-of-the-art stem cell culture systems, with a focus on the recent progress in 3D tissue culture of both embryonic and adult stem cells, as well as the genome editing tools present today. Finally, we will discuss how stem cell and genome editing technologies can be combined to gain insights into human development and disease, and to fulfil the promises of stem cell research in the clinic.
Submitted for review: Aug 26 2015 Published: Dec 10 2015; DOI: 10.18609/cgti.2015.019
Pluripotent stem cells & organoid culture
Since the existence of pluripotent stem cells (PSCs) was definitively proven, there have been many groundbreaking achievements. Principally, three main steps have advanced the field. First is the establishment of mouse and human embryonic stem cell (ESC) cultures. In 1981, Evans and Martin derived mouse ESCs (mESCs), which retained pluripotency through culture on feeder cells, from the inner cell mass of the mouse blastocyst [5,6]. Leukemia inhibitory factor (LIF) was then identified as a key factor for maintaining pluripotency during long-term, feeder-free culture of mESCs [7,8]. Human ESCs (hESCs) were derived a decade later [9], and in 2014, two groups reported conditions allowing the culture of naïve hESCs closely resembling mouse naïve cells [10,11].
The second achievement addressed most ethical concerns surrounding the use of fertilized human embryos to generate pluripotent human cells. In 2006, Professor Shinya Yamanka generated induced pluripotent stem cells (iPSCs) – an equivalent of ESCs – from mouse somatic cells by inducing the expression of four transcription factors: Oct4, Sox2, Klf4 and c-Myc [12]. A year later, the successful generation of human iPSCs was reported [13]. Today, iPSCs can be generated from many different species, and a series of protocols for the differentiation of ESCs and iPSCs into a diverse range of specific cell types has been established [14].
The third major advance was the establishment of stem cell-derived 3D organ cultures – often called organoid cultures. Organoids can be generated either from PSCs or adult stem cells (AdSCs), and are generally defined as 3D, self-organizing cellular structures fully or partially resembling their in vivo counterpart in function as well as in cell type composition [15,16]. Pioneered by the groups of Professors Yoshiki Sasai and Hans Clevers, the first organoid cultures were created almost 7 years ago by generating self-organizing cortical tissues from ESC-derived 3D aggregates [17,18]. Shortly thereafter, the adult intestinal stem cell was also utilized to generate a self-organizing epithelial structure [19]. A defined combination of extracellular matrices, chemicals and growth factors mimicking the in vivo niche has enabled a growing list of 3D organ cultures to be established from both PSCs and AdSCs. From PSCs, the list includes organ buds/organoids of retina [20], pituitary [21], cerebrum [22], ureteric bud [23], small intestine [24], thyroid [25], stomach [26] and liver [27]; whilst from AdSCs organoids have been formed from small intestine [19,28], colon [29], liver [30], prostate [31], pancreas [32,33], stomach [34] and lung [35]. The establishment of these 3D cultures has provided a novel platform for studying stem cell behavior, tissue patterning and organ formation in a petri dish. Moreover, these organoids open new avenues for regenerative and personalized medicine. Patient-derived or human iPSC-derived organoids may act as a transplantable cell source and/or as a system in which rapid drug screening can be performed [36,37].
Genome editing tools
In 1987 Professor Mario Capecchi published a method for homologous recombination (HR) in mESCs. HR is dependent on homologous template DNA. Normally this is the sister chromatid; however the introduction of an exogenous DNA template (a targeting vector) has allowed researchers to introduce specific, site-directed mutations or insertions [38]. In these targeting vectors the DNA to be inserted/edited e.g., a tag or loxP site(s), is flanked by DNA sequences homologous to the genomic insertion site. HR whilst very efficient in mESCs, was found to occur with an extremely low frequency in other mammalian systems. The solution, based on the pioneering work of Dr Maria Jasin, was to introduce double strand breaks (DSBs) into a desired site of the genomic DNA to facilitate HR-mediated repair of the DNA [39,40]. This strategy is applied in the three genome engineering tools described below.
The earliest technology involved zinc finger (ZF) nucleases (ZFNs), enzymes which consist of a DNA-binding ZF domain, which binds to DNA with a specificity of 9–18 base pairs (bp), and the DNA-cleaving domain of the FokI restriction endonuclease [41]. The first use of the ZF–FokI fusion protein came in 1996, and 7 years later it was used for HR-mediated gene targeting in human cells [42]. Transcription activator-like effector nucleases (TALENs) are an alternative technology that enables genome editing [43,44]. Both ZFNs and TALENs are designed in pairs, since the FokI domain needs to dimerize in order to cleave the DNA. Whereas the ZFN DNA-binding domain consists of 3–6 ZFs each recognizing a 3 bp sequence, the TALEN DNA-binding domain is composed of several modules of tandem repeats consisting of 33–35 amino acids, each recognizing 1 bp of genomic DNA. To generate new ZFNs/TALENs, modules of known specificity are combined. However, the specificity of the individual ZF modules are affected by interactions between different modules, a phenomenon called context dependency. As a result the TALEN DNA-binding domain is both more modular and easier to design compared to the ZFN [45–48].
The most recently introduced technology is the clustered interspersed palindromic repeat (CRISPR)/CRISPR-associated (Cas) system. Unlike the two previous technologies, the endonuclease is not fused to a DNA-binding domain, and is instead guided to the genomic sequence of interest by a guide RNA sequence (gRNA) to generate a DSB. Designing a gRNA for CRISPR genome editing is both faster and easier than the generation of a functional pair of ZFNs or TALENs. Designing either of the latter requires extensive knowledge of molecular cloning and protein engineering, while the gRNAs can be ordered as oligonucleotides. This has allowed genome-wide loss-of-function screens using a CRISPR/Cas library to be performed in mouse diploid and haploid ESCs, as well as hESCs [49–51]. Since a gRNA library can be constructed directly with synthesized oligonucleotides (target sequence), the candidate genes can be identified by next-generation sequencing of the oligonucleotide part of gRNAs that serve as barcodes. This technique creates new possibilities for identifying novel gene functions in an unprecedentedly rapid manner (reviewed in [52,53]). The three technologies all have their advantages and disadvantages (reviewed in [54,55] and by Andrew Bassett within this Spotlight issue). Considering the rapid increase in publications using genome editing in recent years, CRISPR/Cas is currently the most popular option, though off-target effects will still need to be minimized if this technique is to be used in the clinic [56–58].
Coupling stem cell & organoid culture with genome editing
Genome editing tools have been widely utilized in stem cell and organoid cultures, and this article will focus on some specific examples. Precise genome editing through DSBs makes it possible to generate a disease-related mutation, thus generating isogenic stem cell lines where ideally the parental cell line has the same genotype, with the exception of the modified site; however, clonal heterogeneity acquired in culture still makes this challenging. Using ZFNs, Soldner et al introduced a dominant mutation strongly linked to familial Parkinson’s disease (A53T mutation) into the α-synuclein gene in healthy donor-derived hESCs, elegantly demonstrating the use of genetic modifications in isogenic cell lines for disease modelling [59]. CRISPR/Cas tools and organoid culture have also been used for in vitro tumor modelling. Intestinal organoids derived from human donors underwent genome editing using the CRISPR/Cas system to introduce known cancer-causing mutations into the four genes (KRAS, APC, TP53 and SMAD4) that are most frequently mutated in colorectal cancer. The resultant mutant organoids could grow independently of all stem cell niche factors, and when xenotransplanted into mice the quadruple mutant organoids were tumorigenic and displayed features of invasive adenocarcinoma [28,60].
Future of monogenic disorders
Applying gene editing technologies to PSC cultures and derivatives allows the development of promising autologous cell sources for transplantation with unlimited expansion capacity. As discussed above, disease models can be generated by the introduction of mutations into healthy donor material. The opposite can therefore be achieved using diseased material: instead of introducing mutations, the mutation(s) causing the disease can be corrected using genome editing (Figure 1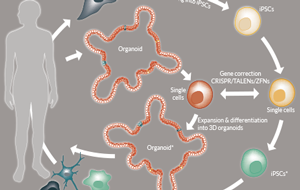
This milestone was achieved first in PSCs and now also in AdSC-derived organoids. In PSCs the first gene to be corrected was a mutant form of IL2GR, which causes X-linked severe combined immunodeficiency [61,62]. Schwank et al repaired the disease-causing mutation in intestinal organoids derived from cystic fibrosis patients. Cystic fibrosis is an autosomal recessive disorder where both copies of the cystic fibrosis transmembrane conductance receptor (CFTR) are mutated. CFTR is an ion channel and its dysfunction results in the disturbed transport of fluid and thickening of the mucus in organs such as the lung, pancreas and small intestine [63,64]. Patient-derived organoids fail to swell in a functional assay involving application of the small molecule Forskolin. However, following HR-mediated gene correction using CRISPR/Cas, the gene-corrected patient organoids performed as well as healthy control organoids in the Forskolin swelling assay [65]. Furthermore, the CFTR mutation has also been corrected in human iPSCs [66]. Although the precise gene correction of CFTR in PSCs and AdSC-derived culture is a great achievement and provides a proof-of-concept showing that organoids and iPSCs hold great potential for cell therapy, it is important to note that in order to be ‘cured’ the patient would need entire tissue replacement using the gene-corrected cells in several tissues and rather large surface areas. At the moment this still represents a large technical obstacle to overcome. However, diseases where the functional complication is restricted to a certain tissue or cell type and is caused by one gene could still be considered as a relatively easy target for gene-edited cell therapy. Potential candidates for current technologies are presented below.
Alpha 1 anti-trypsin deficiency
Alpha 1 anti-trypsin (A1AT) deficiency is an autosomal co-dominant hereditary disorder caused by an inactivating mutation in the A1AT gene. In healthy individuals, A1AT functions as a protease inhibitor and protects tissues primarily by inhibiting enzymes secreted by inflammatory cells. Reduction or lack of function of A1AT results in chronic tissue degradation, mainly of the lung. In addition, certain mutations can cause the misfolding and improper secretion of the protein, causing damage to the liver and eventually leading to liver cirrhosis [67,68]. Due to its liver-specific expression and monogenic cause, A1AT deficiency is a potential target for gene-edited cell therapy. Gene correction has already been performed in iPSCs using ZFNs [69].
Familial hypercholesterolemia (ApoB & PCSK9)
Hypercholesterolemia is defined by elevated levels of cholesterol in the blood which later lead to atherosclerosis and subsequently cardiovascular disease [70,71]. The enzyme proprotein convertase subtilisin type 9 (PCSK9) is mutated in familial hypercholesterolemia, which is an autosomal dominant disease. PCSK9 is expressed in the liver, where it induces degradation of the low-density lipoprotein receptor (LDLR), resulting in a reduction of the rate of degradation of low-density lipoprotein (LDL) cholesterol. Mutations causing PCSK9 to bind more efficiently to the LDLR receptor consequently result in a higher level of LDL in the blood. Due to its inhibitory function (on LDL degradation) the presence of mutant PCSK9 is sufficient to cause the disease. Therefore, while the disease will be partially alleviated following the introduction of gene-corrected cells, the presence of remaining mutant cells may cause some symptoms to persist. On the other hand, Apolipoprotein B (ApoB) binds lipids (e.g., LDL) as well as LDLR, leading to clearance of the lipid. Mutations (e.g., R3500Q) causing ApoB loss-of-function decrease LDL degradation and consequently lead to elevated LDL levels [72,73]. In this case, introduction of gene-corrected cells producing a functional form of the protein may be enough to fully eliminate disease symptoms. Antisense nucleotides targeting PCSK9 have been shown to lower LDL in non-human primates. In addition, in vivo genome editing of PCSK9 using CRISPR/Cas resulted in reduction of cholesterol levels in mice, showing that introduced inactivating mutations can have beneficial effects [71,74–77]. Cell therapy may still be a valuable alternative in the future.
For organoids and PSCs to be used in clinical practice, safe transplantation back to the original patient is essential and this poses a challenge. Although gene-corrected iPSCs can be differentiated into the cell type of interest, contaminating pluripotent cells may cause tumors in the recipient [78–80]. Removal of the remaining PSCs following differentiation is therefore crucial. AdSC-derived organoids have been shown to be genetically stable and the tissue identity of these organoids also seems to be stable unless challenged by genetic modifications [81,82]. However, careful examination of tumorigenic or other malfunctions of AdSC or PSC-derived organoids is still required.
Summary
The last two decades have brought significant improvements to both stem cell culture systems and genome editing tools. PSC- or AdSC-derived 3D culture systems have enabled new ways of modelling tissue patterning, organ formation and complex diseases such as cancer in an unprecedented manner. Meanwhile CRISPR/Cas technology has made gene editing simple and widely available for researchers working in various fields and model systems. Together, these two revolutionary technologies – stem cell-derived organoid cultures and genome editing tools – will bring us one step closer to fulfilling the dream of many stem cell biologists today: applying gene-edited cell therapy to routine medical practice.
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
ACKNOWLEDGMENTS:
The authors would like to thank Chris Hindley and Lawrence Bates for critical reading of the manuscript. Koo BK is supported by the Sir Henry Dale Fellowship from the Wellcome Trust and Andersson-Rolf A is supported by the Medical Research Council (MRC).

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1. Lajtha LG. Stem Cell Concepts, Differentiation 1979; 14(1–3), 23–33. CrossRef
2. Ramalho-Santos M & Willenbring H. On the Origin of the Term ‘Stem Cell.’ Cell Stem Cell 2007; 1(1), 35–8.CrossRef
3. Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell 1997; 88(3), 287–98.CrossRef
4. Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell 2000; 100(1), 157–68.CrossRef
5. Evans MJ & Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981; 292(5819), 154–6. CrossRef
6. Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl Acad. Sci. 1981; 78(12), 7634–8.CrossRef
7. Smith AG, Heath JK, Donaldson DD et al. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides., Nature 1988; 336(6200), 688–90.CrossRef
8. Williams RL, Hilton DJ, Pease S et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988; 336(6200), 684–7.CrossRef
9. Thomson JA. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998; 282(5391), 1145–7.CrossRef
10. Takashima Y, Guo G, Loos R et al. Resetting Transcription Factor Control Circuitry toward Ground-State Pluripotency in Human. Cell 2014; 158(6), 1254–69.CrossRef
11. Theunissen T, Powell B, Wang H et al. Systematic Identification of Culture Conditions for Induction and Maintenance of Naive Human Pluripotency. Cell Stem Cell 2014; 15(4), 471–87.CrossRef
12. Takahashi K & Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126(4), 663–76.CrossRef
13. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131(5), 861–72.CrossRef
14. Stadtfeld M & Hochedlinger K. Induced pluripotency: history, mechanisms and applications. Genes Dev. 2010; 2239–23.CrossRef
15. Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell 2013; 154(2), 274–84.CrossRef
16. Lancaster MA & Knoblich JA. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014; 345(6194), 1247125.CrossRef
17. Eiraku M, Watanabe K, Matsuo-Takasaki M et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008; 3(5), 519–32.CrossRef
18. Lancaster AM, Renner M, Martin C-A et al. Cerebral organoids model human brain development and microcephaly. Nature 2013; 501(7467), 373–9.CrossRef
19. Sato T, Vries RG, Snippert HJ et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009; 459(7244), 262–5.CrossRef
20. Nakano T, Ando S, Takata N et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012; 10(6), 771–85.CrossRef
21. Suga H, Kadoshima T, Minaguchi M et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature 2011; 480(7375), 57–62.CrossRef
22. Lancaster MA, Renner M, Martin C-A et al. Cerebral organoids model human brain development and microcephaly. Nature 2013; 501(7467), 373–9.CrossRef
23. Xia Y, Nivet E, Sancho-Martinez I et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat. Cell Biol. 2013; 15(12), 1507–15.CrossRef
24. Spence JR, Mayhew CN, Rankin SA et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011; 470(7332), 105–9.CrossRef
25. Antonica F, Kasprzyk DF, Opitz R et al. Generation of functional thyroid from embryonic stem cells. Nature 2012; 491(7422), 66–71.CrossRef
26. McCracken KW, Catá EM, Crawford CM et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014; 516(7531), 400–4.CrossRef
27. Takebe T, Sekine K, Enomura M et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013; 499(7459), 481–4.CrossRef
28. Drost J, van Jaarsveld RH, Ponsioen B et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015; 521(7550), 43-7.CrossRef
29. Yui S, Nakamura T, Sato T et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat. Med. 2012; 18(4), 618–23.CrossRef
30. Huch M, Dorrell C, Boj SF et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013; 494(7436), 247–50.CrossRef
31. Karthaus WR, Iaquinta PJ, Drost J et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014; 159(1), 163–75.CrossRef
32. Huch M, Bonfanti P, Boj SF et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013; 32(20), 2708–21.CrossRef
33. Boj SF, Hwang C-I, Baker LA et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2014; 160(1–2), 324–38. CrossRef
34. Bartfeld S, Bayram T, van de Wetering M et al. In vitro Expansion of Human Gastric Epithelial Stem Cells and Their Responses to Bacterial Infection. Gastroenterology 2014; 148(1), 126–36.CrossRef
35. Lee J-H, Bhang DH, Beede A et al. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014; 156(3), 440–55.CrossRef
36. Sterneckert JL, Reinhardt P, Schöler HR. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014; 15(9), 625–39.CrossRef
37. Rookmaaker MB, Schutgens F, Verhaar MC, Clevers H. Development and application of human adult stem or progenitor cell organoids. Nat. Rev. Nephrol. 2015; 11(9), 546–54.CrossRef
38. Thomas KR & Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987; 51(3), 503–12. CrossRef
39. Rouet P, Smih F, Jasin M, Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl Acad. Sci. USA 1994; 91(13), 6064–8.CrossRef
40. Jasin M & Rothstein R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2013; 5(11), a012740.CrossRef
41. Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA 1998; 95(18), 10570–75.CrossRef
42. MH Porteus & Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003; 300(5620), 763.CrossRef
43. Moscou MJ & Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science 2009; 326(5959), 1501.CrossRef
44. Boch J, Scholze H, Schornack S et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009; 326(5959), 1509–12.CrossRef
45. Isalan M, Choo Y, Klug A. Synergy between adjacent zinc fingers in sequence-specific DNA recognition. Proc. Natl Acad. Sci. USA 1997; 94(11), 5617–21.CrossRef
46. Isalan M, Klug A, Choo Y. Comprehensive DNA recognition through concerted interactions from adjacent zinc fingers. Biochemistry 1998; 37(35), 12026–33.CrossRef
47. DeFrancesco L. Move over ZFNs. Nat. Biotechnol. 2011; 29(8), 681–4.CrossRef
48. Sander JD, Dahlborg EJ, Goodwin MJ et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat. Methods 2011; 8(1), 67–9.CrossRef
49. Leeb M, Dietmann S, Paramor M, Niwa H, Smith A. Genetic exploration of the exit from self-renewal using haploid embryonic stem cells. Cell Stem Cell 14(3), 385–93.CrossRef
50. Mali P, Yang L, Esvelt KM et al. RNA-guided human genome engineering via Cas9. Science 2013; 339(6121), 823–6.CrossRef
51. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014; 343(6166), 80–4.CrossRef
52. Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015; 16(5), 299–311.CrossRef
53. Agrotis A & Ketteler R. A new age in functional genomics using CRISPR/Cas9 in arrayed library screening. Front. Genet. 2015; 6, 300.CrossRef
54. Kim H & Kim J-S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014; 15(5), 321–34.CrossRef
55. Vasileva EA, Shuvalov OU, Garabadgiu AV, Melino G, Barlev NA. Genome-editing tools for stem cell biology. Cell Death Dis. 2015; 6(7), e1831.CrossRef
56. Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013; 31(9), 839–43.CrossRef
57. Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013; 41(20), 9584–92.CrossRef
58. Fu Y, Foden JA, Khayter C et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31(9), 822–6.CrossRef
59. Soldner F, Laganière J, Cheng AW et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011; 146(2), 318–31.CrossRef
60. Matano M, Date S, Shimokawa M et al. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015; 21(3), 256–62.CrossRef
61. Urnov FD, Miller JC, Lee Y-L et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005; 435(7042), 646–51.CrossRef
62. Firth AL, Menon T, Parker GS et al. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015; 12(9), 1385–90.CrossRef
63. O’Sullivan BP & Freedman SD. Cystic fibrosis. Lancet 2009; 373(9678), 1891–1904.CrossRef
64. Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006; 440(7083), 477–83.CrossRef
65. Schwank G, Koo B-K, Sasselli V et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013; 13(6), 653–8.CrossRef
66. Crane AM, Kramer P, Bui JH et al. Targeted Correction and Restored Function of the CFTR Gene in Cystic Fibrosis Induced Pluripotent Stem Cells. Stem Cell Reports 2015; (4), 569–77.CrossRef
67. Fairbanks KD & Tavill AS. Liver disease in alpha 1-antitrypsin deficiency: a review. Am. J. Gastroenterol. 2008; 103(8), 2136–41.CrossRef
68. Stoller JK & Aboussouan LS, Alpha1-antitrypsin deficiency. Lancet 2005; 365(9478), 2225–36.CrossRef
69. Yusa K, Rashid ST, Strick-Marchand H et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature 2011; 478(7369), 391–4.CrossRef
70. Durrington P. Dyslipidaemia. Lancet 2003; 362(9385), 717–31.CrossRef
71. Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J. Clin. Invest. 2003; 111(12), 1795–803.CrossRef
72. Myant N. Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolaemia. Atherosclerosis 1993; 104(1–2), 1–18.CrossRef
73. Innerarity TL, Mahley RW, Weisgraber KH et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J. Lipid Res. 1990; 31(8), 1337–49.CrossRef
74. Lambert G, Sjouke B, Choque B, Kastelein JJ P, Hovingh GK. The PCSK9 decade. J. Lipid Res. 2012; 53, 2515–24.CrossRef
75. Abifadel M, Varret M, Rabès J-P et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003; 34(2), 154–6.CrossRef
76. Lindholm MW, Elmén J, Fisker N et al. PCSK9 LNA Antisense Oligonucleotides Induce Sustained Reduction of LDL Cholesterol in Nonhuman Primates. Mol. Ther. 2012; 20(2), 376–81.CrossRef
77. Ran FA, Cong L, Yan WX et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015; 520(7546), 186-91.CrossRef
78. Lu X & Zhao T. Clinical Therapy Using iPSCs: Hopes and Challenges, Genomics. Proteomics Bioinformatics 2013; 11(5), 294–8.CrossRef
79. Ben-David U & Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011; 11(4), 268–77.CrossRef
80. Ghosh Z, Huang M, Hu S, Wilson KD, Dey D, Wu JC. Dissecting the oncogenic and tumorigenic potential of differentiated human induced pluripotent stem cells and human embryonic stem cells. Cancer Res. 2011; 71(14), 5030–9.CrossRef
81. Huch M, Gehart H, Van Boxtel R et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015; 160(1–2), 299–312.CrossRef
82. Simmini S, Bialecka M, Huch M et al. Transformation of intestinal stem cells into gastric stem cells on loss of transcription factor Cdx2. Nat. Commun. 2014; 5, 5728.CrossRef
Affiliations
Amanda Andersson-Rolf
Department of Genetics and Wellcome Trust – Medical Research Council Stem Cell Institute, the University of Cambridge
Cambridge, United Kingdom; ama66@cam.ac.uk
Corresponding author
Bon-Kyoung Koo
Department of Genetics and Wellcome Trust – Medical Research Council Stem Cell Institute, the University of Cambridge, Cambridge, United Kingdom bkk25@cam.ac.uk