
A US Cell Therapy Regulatory Toolkit
Cell Gene Therapy Insights 2015; 1(1), 129-132.
10.18609/cgti.2015.012
Emily J Culme-Seymour, James Lawford Davies, Julian Hitchcock, Julian Mason, Melissa K Carpenter & Chris Mason Navigating the Global Regulatory Landscape The successful translation of cell and gene therapies into the clinic requires careful navigation of the regulatory pathways during product development and commercialization. The US Cell Therapy Regulatory Toolkit has been developed to inform […]
Submitted: August 12 2015 Published: September 15 2015;
Steps to product
The first section covers the various meetings required by FDA in preparation for an investigational new drug (IND) application, including what those meetings will entail, specific details on timings and logistics, and briefing document requirements (Figure 1
Manufacturing
The second section looks at the manufacturing requirements for each stage of product development from preclinical development up to commercialization (Figure 2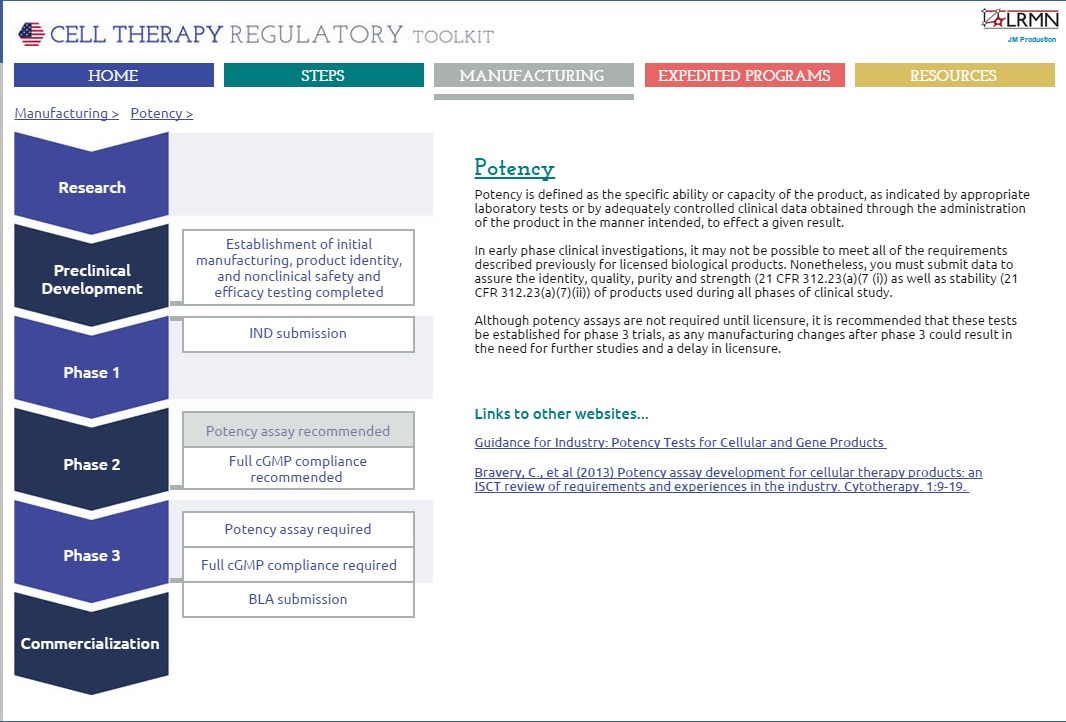
Expedited Programs
The third section details the various expedited programmes available to those developing cell therapies in the US (Figure 3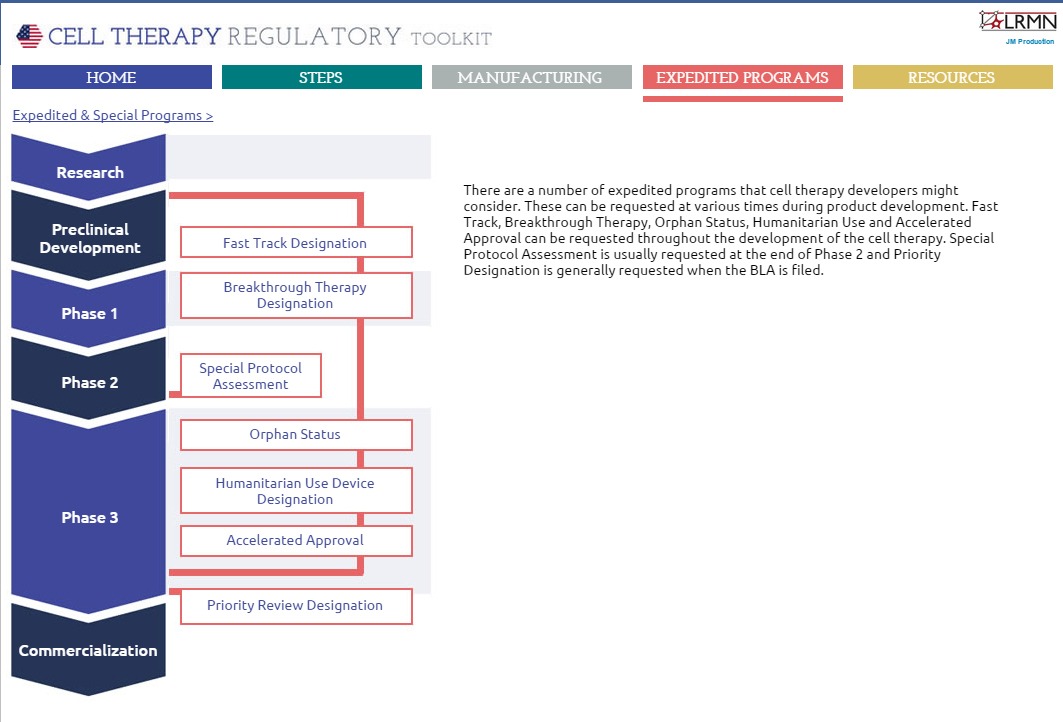
Conclusion
The information contained within the US Cell Therapy Regulatory Toolkit provides accessible regulatory education to users about specific requirements they can expect during product and clinical development in the USA. Users are welcome to navigate the pages, and use it as a starting point in seeking to understand the regulatory pathway for taking cell therapies into the clinic.
Endnote
The toolkit was constructed using information available in 2014. It is inevitable that regulatory requirements will change over time, and looking ahead, users should therefore check whether there have been any subsequent changes introduced by the relevant regulatory bodies.
Acknowledgements
The authors are grateful to Karen Rowland for design and creative concept for the toolkit.
Financial & competing interests disclosure
The Cell Therapy Regulatory Toolkit was developed within the British Regen Industry Tool Set (BRITS) project (2010–2014) funded by Innovate UK (then the Technology Strategy Board) under their Regenerative Medicine Program: Value Systems and Business Modelling. C Mason was the Principle Investigator and EJ Culme-Seymour was an investigator on the project.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
Affiliations
Emily Culme-Seymour
Director, London Regenerative Medicine Network, 2 Royal College Street, London NW1 0NH, UK
James Lawford Davies
Hempsons, Hempsons House, 40 Villiers Street, London WC2N 6NJ, UK
Julian Hitchcock
Denoon Legal, 14a Clerkenwell Green, London EC1R 0DP, UK
Julian Mason
Faculty of Science, Engineering and Computing, Kingston University, Penrhyn Road, Kingston upon Thames, Surrey KT1 2EE, UK
Melissa K Carpenter
Carpenter Group Consulting Inc., Washington, USA
Chris Mason
Professor of Regenerative Medicine Bioprocessing, Advanced Centre for Biochemical Engineering, University College London, Bernard Katz Building, Gordon Street, London WC1H 0AH, UK